Article Text

Abstract
Objective We aimed to carry out ocular examination and genetic studies in a family in which some members are affected with osteogenesis imperfecta (OI) and primary open-angle glaucoma (POAG). We compared the corneal properties of affected and unaffected members (ie, cases and controls).
Methods Eight family members from two generations, both affected and unaffected, were examined. Corneal hysteresis (CH), intraocular pressure (IOP) measured with Goldmann applanation tonometer, central corneal thickness (CCT) and cornea-corrected IOP (IOPcc) were recorded. Blood samples were obtained from seven family members, both affected and unaffected, and tested for a panel of genes associated with OI.
Results Family members affected with OI (n=6) had a heterozygous splice site mutation in intron 26 of the COL1A1 gene. The family members affected with OI had reduced CCT (476.5±24.6 µm) and CH (7.9 ±1.4 mmHg) compared with the unaffected controls (CCT, 575.8±10.8 µm; CH, 12.3±0.8 mmHg). Two of the six patients affected with OI had a glaucoma diagnosis and were on topical therapy and under regular clinical review.
Conclusions Patients affected with OI have a significant risk of developing POAG due to the effects of abnormal collagen on various ocular structures. Two of these effects which place them at risk are reduced CCT and CH. They should be screened and monitored for glaucoma from a young age, and the examination should include corneal biomechanical measurements and CCT to identify those most at risk. IOPcc may be a more accurate way to monitor IOP in the presence of abnormal corneal properties.
- glaucoma
- intraocular pressure
- cornea
Data availability statement
All data relevant to the study are included in the article.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Patients with osteogenesis imperfecta (OI) have reduced central corneal thickness (CCT) and corneal hysteresis (CH).
Glaucoma is associated with reduced CCT and CH.
OI is associated with the development of glaucoma.
What are the new findings?
Our study shows a difference in CCT and CH between affected and unaffected members of a family with genetically identified OI, some of whom are also affected with glaucoma.
How might these results change the focus of research or clinical practice?
Corneal hysteresis is a non-invasive screening tool and should be integrated into a screening eye examination to be carried out on patients with OI at risk of developing glaucoma.
Introduction
Osteogenesis imperfecta (OI) or ‘brittle bone disease’ is a genetically heterogeneous skeletal dysplasia that affects approximately 1 in 10 000–20 000 births.1 The spectrum of skeletal abnormalities varies from low bone mass seen with OI type I (70% of all cases), to progressive bone deformities with frequent fractures (OI type III/IV), to perinatal lethality (OI type II).2 There can be variable dentinogenesis imperfecta and, in adult years, hearing loss.
Manschot first described the ocular abnormalities associated with OI based on a postmortem examination of a baby with type II OI, namely, a markedly thin cornea and sclera (giving rise to the appearance of ‘blue sclera’), along with abnormally deep excavation of the optic disc, ‘probably caused by insufficient development of the lamina cribrosa’.3 Other associated ocular abnormalities that have been described include arcus senilis, small corneal diameter, small globe length, myopia, secondary glaucoma, optic atrophy, retinal detachment, subhyaloid haemorrhage, vitreous hyperplasia, congenital absence of Bowman’s layer, megalocornea, corneal opacities and keratoconus.4
More recently, an association has been made by Wallace et al between OI and primary open-angle glaucoma (POAG). They identified two novel mutations in the COL1A1 gene in individuals with OI type I and POAG, and concluded that some COL1A1 mutations may be causative for both OI and POAG.5 Mauri et al further suggested collagen mutation screening be included in the genetic workup of early-onset glaucoma cases and detailed ophthalmological examinations be carried out on all patients with OI .6
We report on eight subjects from two generations of a family in which some members are affected with type I OI and some with POAG. To the best of our knowledge, ours is the first study of its type looking at corneal properties (including central corneal thickness (CCT) and corneal hysteresis (CH)) of both affected and unaffected family members. Both CCT and CH are associated with POAG.7–11 We aimed to ascertain which genetic mutation was present in this family. We also aimed to establish whether there was a difference in corneal properties between affected and unaffected members.
Methods
Ocular examination
Informed consent was obtained from all subjects. Eight subjects from two generations of the same family (figure 1) underwent a complete medical history and ocular examination. Ocular examination in all subjects included visual acuity (VA), slit-lamp examination, intraocular pressure (IOP) measurement with Goldmann applanation tonometer (GAT; Haag-Streit AG, Koeniz, Switzerland) and examination of the posterior fundus including optic nerve and macula. In the proband (subject 1, affected with OI and POAG), the most recent Humphrey visual field Swedish interactive thresholding algorithm (SITA) Fast 24-2 was collected (Humphrey Instruments, Dublin, California, USA). Other subjects had visual field testing carried out only if clinically indicated.
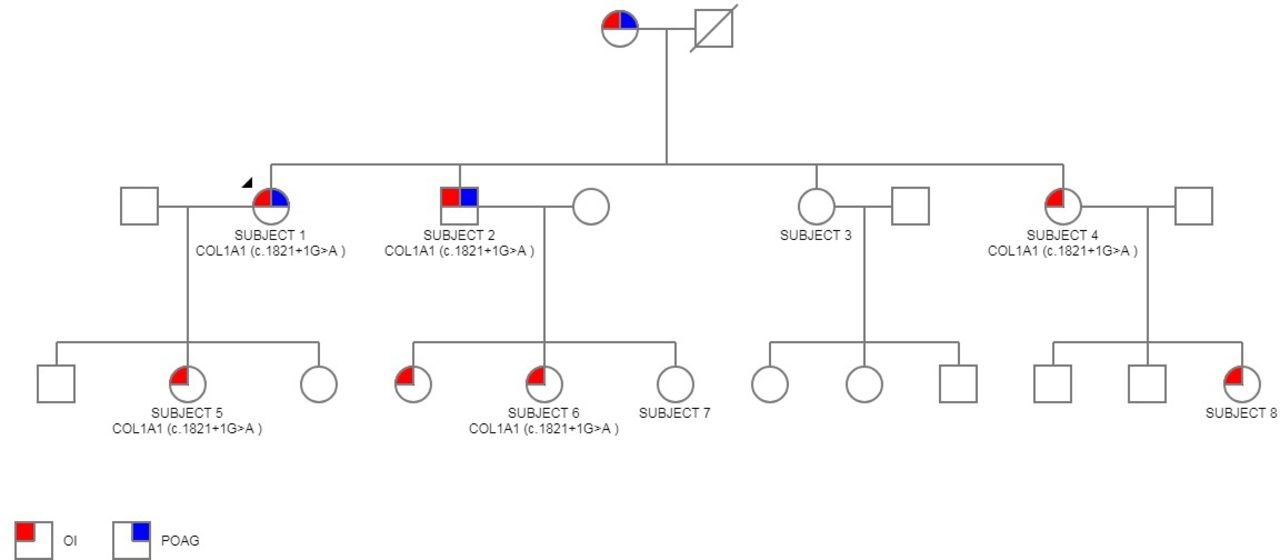
Pedigree chart including eight subjects both affected and unaffected. Proband is subject 1. Genotypes of affected patients as indicated.
Corneal properties were recorded as follows: CCT with ultrasound pachymetry (Pachmate 2; DGH Technology, Philadelphia, Pennsylvania, USA) and CH with the Ocular Response Analyser (ORA; Reichert, Depew, New York, USA). The ORA measures the corneal response to indentation by a rapid air pulse that causes the central cornea to move inwards, past applanation and into slight concavity before returning to normal curvature. This action permits the detection of a second applanation point, as the cornea returns from its overapplanated state. Using the first applanation pressure point (P1) and the second applanation pressure point (P2), Goldmann-correlated IOP (IOPg) is the average of the two. During the process, the cornea absorbs energy from the initial air pulse, which causes the second applanation pressure measurement to be lower than the initial measurement. The difference between the two pressures (P1−P2) is CH. CH is thought to represent the viscoelastic nature of the cornea or its ‘viscous-damping’ capacity.12
P1 and P2 can be combined algorithmically to provide cornea-corrected IOP (IOPcc). The formula was derived by minimising the absolute average IOP difference for eyes before and after laser in situ keratomileusis. In this way, it was validated as having essentially entirely removed corneal effects from IOPcc measurements.13 Subsequent studies on eyes with both normal and thin corneas have shown that IOPcc is less affected by CCT than GAT.14 15 Medeiros and Weinreb suggested that for this reason it may be a more suitable method for IOP monitoring in patients with abnormal corneal properties.15
Note that ‘normal’ CCT varies by study population. The mean CCT in a large cross-sectional European population-based study by Hoffmann et al was 554.2±34.8 µm.16 The mean CH in the cross-sectional cohort UK Biobank study was 10.60±1.88 mmHg.17
Genetic testing
Blood samples were obtained from seven of the eight subjects. The proband (subject 1) sample underwent sequencing of genomic DNA for a panel of 21 genes associated with autosomal-dominant OI (table 1). The remaining six samples underwent targeted sequencing on both DNA strands of the relevant COL1A1 region.
The 21-gene osteogenesis imperfecta panel
Results
Subjects
The eight subjects’ examination results are summarised in table form in table 2.
Summary of subjects’ characteristics
Subject 1
A female in her late 50s with a clinical diagnosis of OI type I made at the age of 36, when her infant daughter (subject 5) had several low-impact long bone fractures. She was diagnosed with POAG at the age of 40, with a presenting IOP of 26 mmHg oculus uterque (OU). She was on two topical IOP-lowering agents: a prostaglandin analogue (PA) and a carbonic anhydrase inhibitor. She also described a recent hearing impairment. On examination, she had an unaided VA of 6/6 OU. Slit-lamp examination revealed bilateral glaucomatous optic disc cupping. IOP GAT was 13 mmHg oculus dextrus (OD) and 18 mmHg oculus sinister (OS). IOPcc was 29 mmHg OD and 21 mmHg OS. CCT was 489 µm OD and 476 µm OS. CH was 8.2 mmHg OD and 9.4 mmHg OS. Humphrey visual field 24-2 showed bilateral glaucomatous visual field defects, with a mean deviation of −13.13 dB OD (figure 2) and −5.32 dB OS. This subject’s mother (aged 83) also had OI POAG and is included in the pedigree chart (figure 1) but was not available for collection of data. She lives in a long-term care facility with poor mobility and visual impairment due to end-stage POAG.

Humphrey visual field test of subject 1, oculus dextrus showing glaucomatous visual field loss. GHT, glaucoma hemifield test; MD, mean deviation; PSD, pattern standard deviation; VFI, Visual Field Index.
Subject 2
A male in his mid-50s with a clinical diagnosis of OI and POAG. He was diagnosed with POAG at another institution at age 45 with a presenting IOP in the mid-20s OU. He was on one topical IOP-lowering agent, a PA, at the time of examination. His ocular history was significant for left amblyopia. On examination, he had a corrected VA of 6/9 OD and 6/18 OS. He had bilateral iris transillumination defects (TIDs) and bilateral glaucomatous optic disc cupping. There were no significant clinical corneal findings. IOP GAT was 24 mmHg OU. IOPcc was 28 mmHg OD and 39 mmHg OS. CCT was 433 µm OD and 447 µm OS. CH was 7.5 mmHg OD and 4.1 mmHg OS.
Subject 3
An unaffected female in her early 50s with no ocular history. On examination, she had an unaided VA of 6/9 OU. Slit-lamp examination revealed bilateral iris TIDs and normal, healthy-looking optic discs. There were no significant clinical corneal findings. IOP GAT was 17 mmHg OU. IOPcc was 19 mmHg OD and 18 mmHg OS. CCT was 589 µm OD and 578 µm OS. CH was 12.6 mmHg OD and 12.1 mmHg OS.
Subject 4
A female in her mid-40s with OI and no ocular history. On examination, she had a corrected VA of 6/9 OU. Slit-lamp examination of anterior segment was unremarkable and she had healthy optic discs with no cupping. IOP GAT was 20 mmHg OD and 23 mmHg OS. IOPcc was 26 mmHg OD and 29 mmHg OS. CCT was 482 µm OD and 476 µm OS. CH was 8.0 mmHg OD and 7.0 mmHg OS. Humphrey visual field SITA Fast 24-2 was within normal limits in both eyes. She was commenced on a PA for ocular hypertension.
Subject 5
A female in her early 20s with OI and no ocular history. On examination, she had an unaided VA of 6/6 OU. Slit-lamp examination was unremarkable. IOP GAT was 16 mmHg OU. IOPcc was 19 mmHg OU. CCT was 476 µm OD and 471 µm OS. CH was 7.7 mmHg OD and 7.8 mmHg OS.
Subject 6
A female in her late teens with OI and no ocular history. On examination, she had an uncorrected VA of 6/5 OU. Slit-lamp examination revealed iris TIDs and healthy optic nerves. There were no significant clinical corneal findings. IOP GAT was 20 mmHg OD and 21 mmHg OS. IOPcc was 25 mmHg OU. CCT was 524 µm OD and 491 µm OS. CH was 9.7 mmHg OD and 8.6 mmHg OS.
Subject 7
An unaffected female in her mid-teens with no ocular history. On examination, she had a corrected VA of 6/6 OU. Slit lamp examination was unremarkable. IOP GAT was 12 mmHg OU. IOPcc was 15 mmHg OD and 22 mmHg OS. CCT was 563 µm OD and 573 µm OS. CH was 13.2 mmHg OD and 11.4 mmHg OS.
Subject 8
A female child with OI and no ocular history. This subject, at her young age, had suffered multiple fractures to date. On examination, she had an unaided VA of 6/6 OD and 6/9 OS. Slit-lamp examination was unremarkable. IOPg (ORA) was 17 mmHg OD and 18 mmHg OS. IOPcc was 19 mmHg OD and 20 mmHg OS. CH was 8.8 mmHg OD and 7.9 mmHg OS. Due to her age, this subject was unable to partake in blood sampling, GAT and pachymetry.
IOP and corneal properties in cases and controls
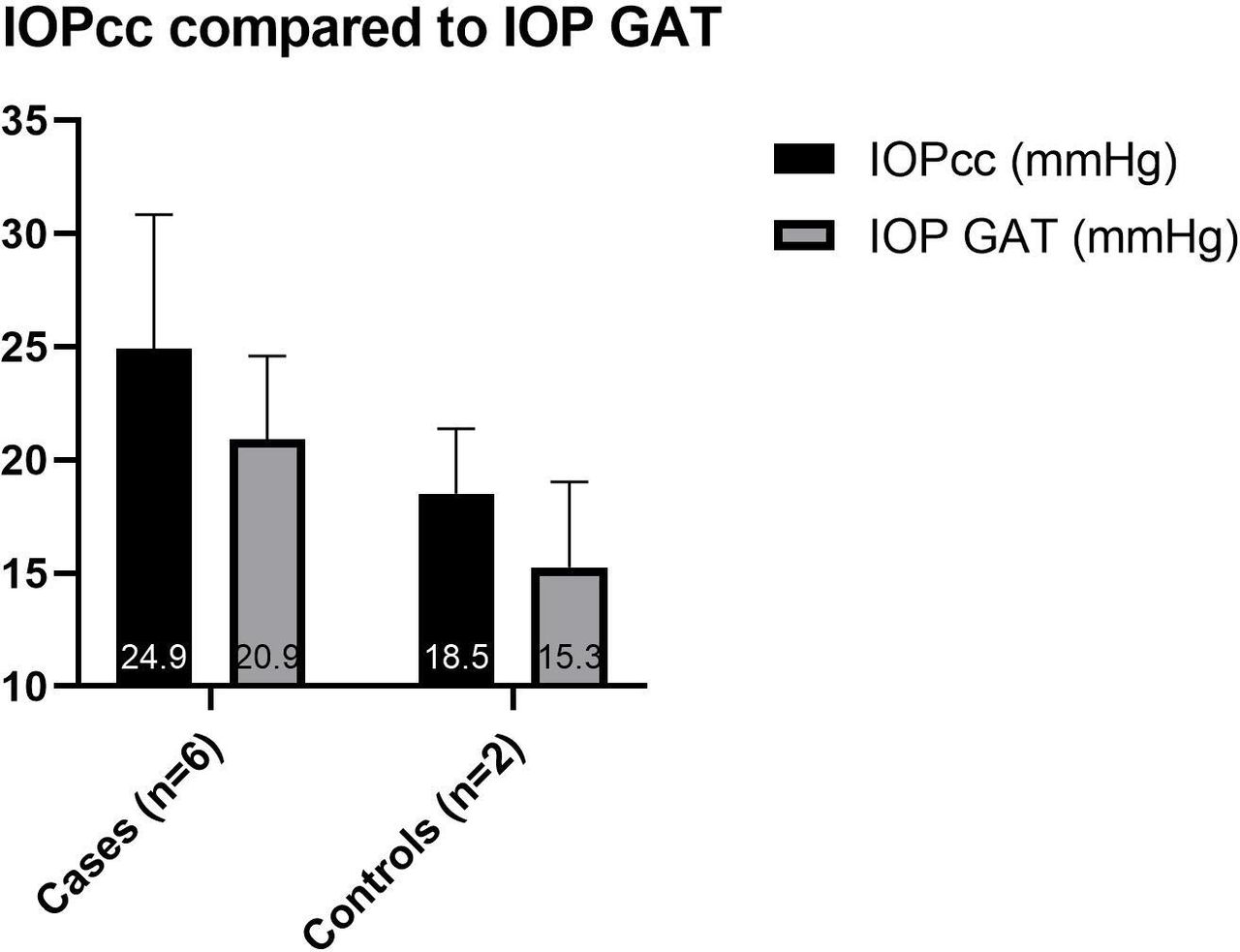
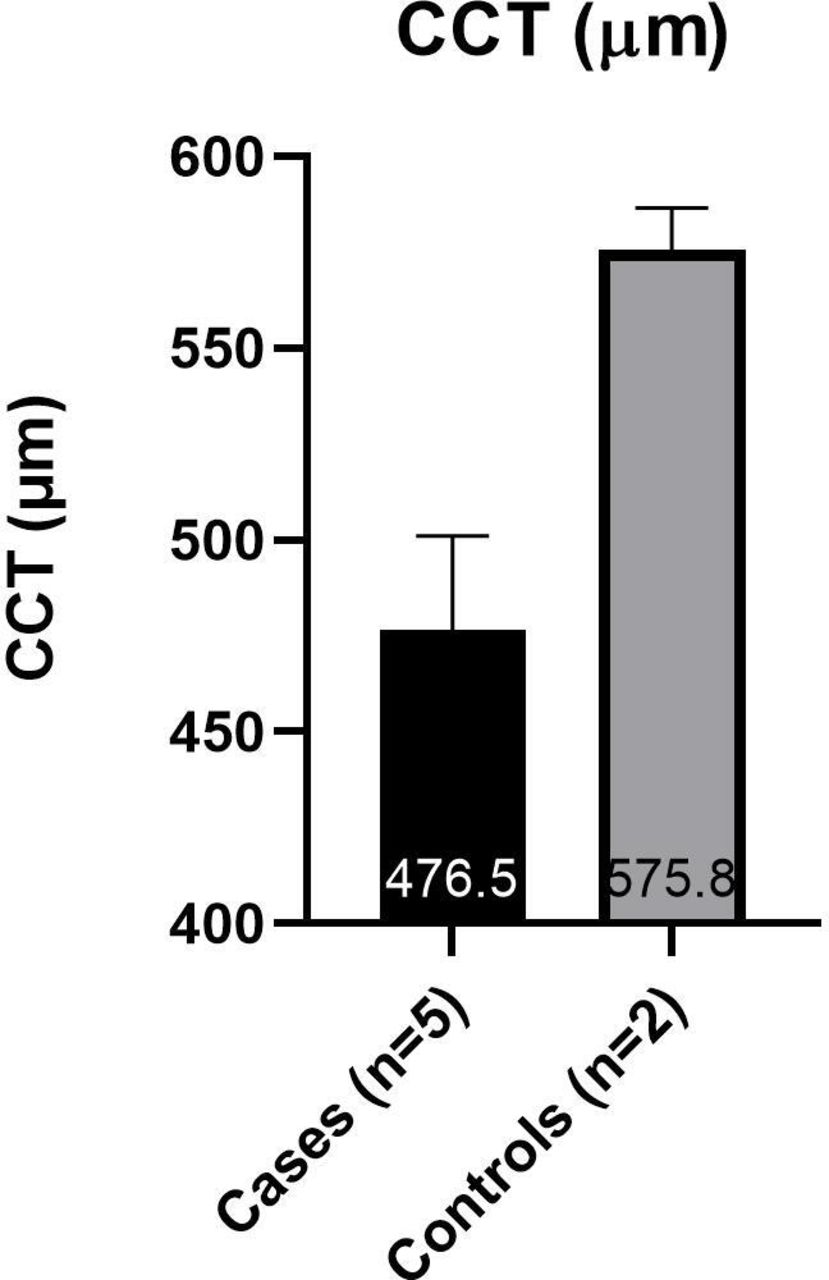
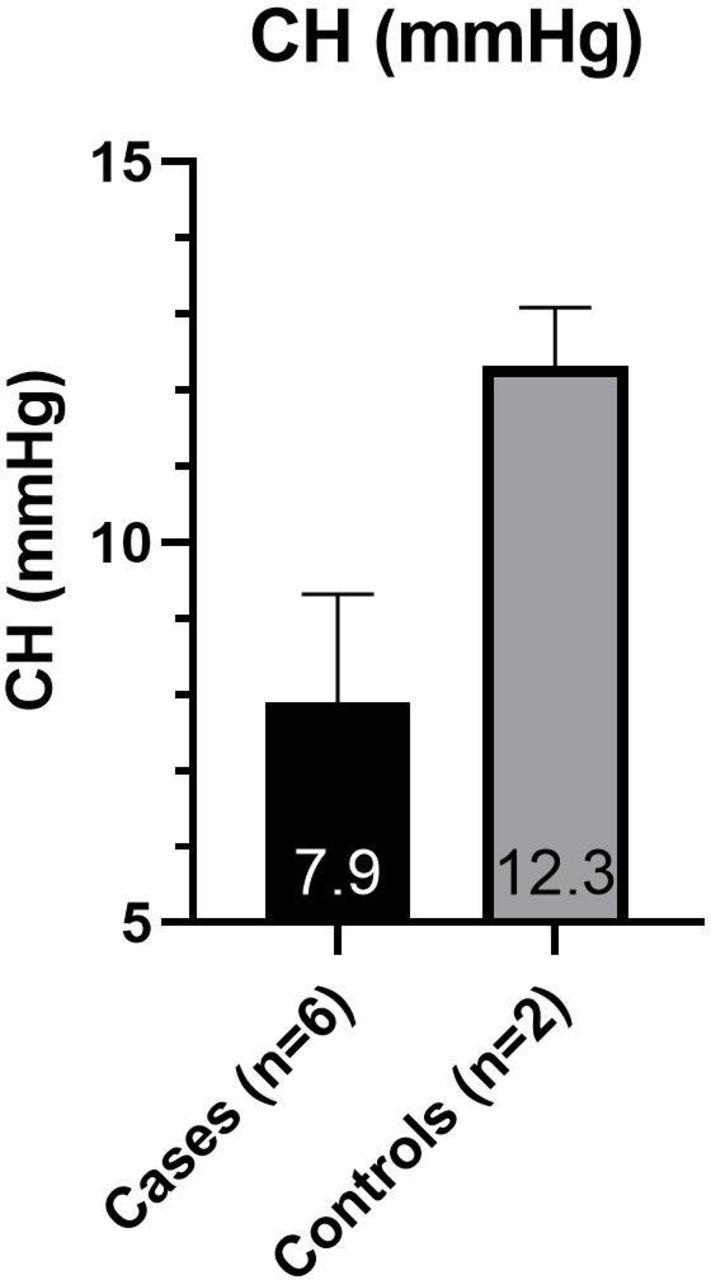
Subjects were arranged into two groups for data analysis. The ‘cases’ group includes those affected with OI; the ‘controls’ group includes those unaffected with OI. The average values from both eyes were used for each subject. Due to the small sample size (cases n=6, controls n=2) and the data being largely abnormally distributed, statistical analysis was not carried out. The mean IOPcc was higher than the mean IOP GAT in all subjects (figure 3). The cases had a higher mean IOP GAT (20.9±3.7 mmHg) and IOPcc (24.9±5.9 mmHg) than the controls (15.3±3.8 mmHg and 18.5±2.9 mmHg, respectively) (figure 3). The cases had a lower mean CCT (476.5 µm±25) and CH (7.9 mmHg ±1.4) than the controls (575.8±11 µm and 12.3±0.8 mmHg, respectively) (figures 4 and 5).

Mean (±SD) of IOP GAT and IOPcc in case and control groups. An average of both eyes of each subject is used. Note that subjects 1 and 2 were on ocular antihypertensive treatment at the time of data collection, and a pretreatment IOP GAT was available for subject 1 only. IOPcc, cornea-corrected intraocular pressure measured with the Ocular Response Analyser; IOP GAT, intraocular pressure measured with Goldmann applanation tonometry.
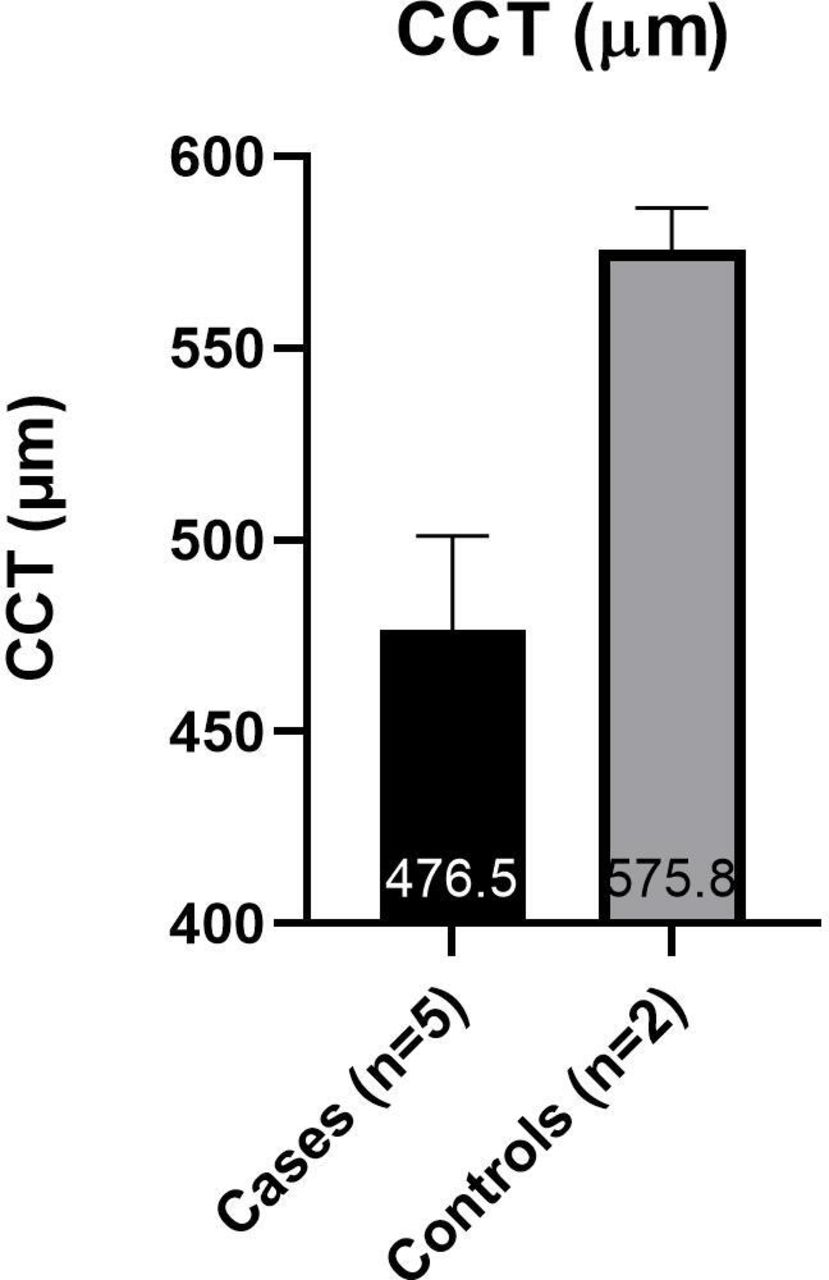
Mean (±SD) of CCT in case and control groups. An average of each subject’s two eyes is used. CCT, central corneal thickness measured by Pachmate 2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (±SD) of CH in case and control groups. An average of each subject’s two eyes is used. CH, corneal hysteresis measured by Ocular Response Analyser.
Genetic testing
Analysis of the family pedigree (figure 1) suggested an autosomal-dominant inheritance pattern. This was confirmed by the finding of a heterozygous c.1821+1G>A splice site mutation in intron 26 of the COL1A1 gene in the subjects affected with OI. The proband (subject 1) underwent 21 gene analysis, and no other mutation or sequence changes were found. The targeted COL1A1 variant was not identified in the two OI-unaffected subjects (subjects 3 and 7).
Discussion
The subjects in OI case group had a mutation of the COL1A1 gene. OI cases had higher IOP (GAT and IOPcc), lower CH and lower CCT than those in the control group. Two of the cases had a known diagnosis of POAG and were on topical therapy and under regular clinical review. Iris TIDs were noted in two of the six patients with OI and one of the unaffected patients. There were no other features of pigment dispersion glaucoma, that is, no Krukenberg’s spindle, dispersed pigment or Zentmayer rings. Iris TIDs are also associated with megalocornea,18 which is a known manifestation of OI. There was no clinical suspicion of megalocornea in any of the subjects.
Genetic mutations in OI
OI was previously described as an autosomal-dominant disorder resulting from mutations in the COL1A1 and COL1A2 genes which code for α1 and α2 chains of type I collagen, respectively. Type I collagen is the most abundant fibrillar collagen of bone, skin and extracellular matrices. In all, 85%–90% of cases of OI are caused by mutations of these two genes. The remainder are recessive forms. Seven types are caused by defects in genes whose protein products interact with collagen for folding or post-translational modifications. Two other rare defects mainly affect bone mineralisation, but also decrease collagen production. The most recently identified genes show primary defects in osteoblast differentiation.1
Collagen mutations may be qualitative (ie, structural) or quantitative. Collagen with a primary structural defect has more severe consequences for intracellular metabolism and matrix structure than does a reduced amount of normal collagen.1 Heterozygous null COL1A1 alleles result in synthesis of a reduced amount (about half) of structurally normal collagen and cause the mildest form of the disorder, OI type I. OI types II–IV commonly are caused by qualitative collagen mutations.19
Our affected subjects have a pathogenic c.1821+1G>A splice site mutation in intron 26 of the COL1A1 gene. Splice site predictive software indicates that this pathogenic mutation would disrupt the donor splice site, leading to abnormal splicing of exon 26.20 This mutation was first reported in an individual with OI type I in 1993 by Stover et al.21 Interestingly, Zhytnik et al later found two unrelated patients with this same mutation. In these two patients, this mutation resulted in phenotypes of different severity: one patient having OI type I and the other having OI type III.22 Genotype−phenotype correlations remain an unresolved issue in our understanding of OI. Cases of interfamilial OI diversity are not rare. Not only genetics but also additional factors, such as epigenetics and environment, might contribute to the development of specific OI phenotypes. Indeed within our group of subjects, all of whom have the same mutation, there is a differing phenotype. Subject 8 is the worst affected, suffering more frequent, disabling and deforming fractures.
Role of collagen in glaucoma
Type I collagen is found in abundance in many ocular tissues, including cornea, sclera, trabecular meshwork (TM) and lamina cribrosa (LC).23 Collagen abnormalities may contribute to the development of glaucoma in a number of ways. Changes in collagen composition of the extracellular matrix of the TM may be responsible for increased outflow resistance and subsequent increased IOP.24 Hernandez et al discussed changes in the connective tissue of the lamina cribrosa in the optic nerve in glaucoma, with an increase in collagen VI and disruption and loss of elastic fibres in the LC playing an important role in the progression of disease.25
Albon et al concluded that mechanical compliance and resilience of the LC decrease with age26 and are associated with differing proportions of collagen subtypes as well as an increase in total collagen.27 These studies suggest an increased susceptibility to permanent deformation with age in the normal population, which may be more significant in those with collagen abnormality.
Glaucoma in animal model COL1A1 mutations
Aihara et al demonstrated elevated IOP in mice with a transgenic mutation in the gene for the α1 subunit of type I collagen. This suggested an association between IOP regulation and fibrillar collagen turnover.28 The same group later reported that aqueous outflow was reduced in mice with this COL1A1 mutation due to increased resistance in the outflow pathway.29 Mabuchi et al used a similar mouse model to demonstrate optic nerve axonal loss (associated with elevated IOP) at 54 weeks of age in those with the targeted COL1A1 mutation.30 These studies suggest that faulty collagen in the TM is one of the mechanisms by which those with COL1A1 mutation are at risk of elevated IOP and subsequent POAG development.
Corneal properties in glaucoma
Reduced CCT is becoming increasingly recognised as an independent risk factor for the development and progression of POAG. In recent studies, reduced CH was a significant predictor of progression in POAG.8–11 In the UK Biobank study, which analysed CH data from 93 345 participants, CH was negatively associated with male sex, age, black ethnicity, self-reported glaucoma, diastolic blood pressure and height. In addition, self-reported glaucoma and CH were significantly associated when CH was less than 10.1 mmHg.17
Corneal properties in human COL1A1 mutations
Dimasi et al analysed the role of mutations in type I collagen genes in CCT and found that CCT was markedly less in patients with OI with mutations in COL1A1 and COL1A2 than matched controls.31 Lagrou et al reported altered corneal properties in children with OI. These children had reduced CCT and CH when compared with age-matched controls.32
CH applications
We can infer information about posterior ocular structures from non-invasive CH measurements. Corneal hysteresis refers to the ability of the cornea to dampen pressure changes, rather than having characteristics of floppiness or rigidity. Wells et al found an association between higher corneal hysteresis values and more optic nerve deformation during acute IOP elevation. They hypothesised that a low CH could correlate with stiffening of the peripapillary sclera and reduced ability to dampen the effects of raised IOP on the optic nerve head.33 Lanzagorta et al, while following up newly diagnosed patients with glaucoma commencing topical treatment, found that the greater the corneal CH at baseline, the greater the LC displacement anteriorly during follow-up, representing recovery to its original position.34 Essentially, eyes with higher hysteresis are better able to recover the original position of the LC when the IOP is lowered.
Study limitations and biases
The major limitation of this study is the small number of subjects. This precluded the possibility of accurate statistical analysis of the data. A potential source of bias came from the fact that subjects 1 and 2 were both on IOP-lowering treatment, which included a PA, at the time of data collection. Pretreatment IOP GAT was used for subject 1, but not for subject 2 as it was unavailable. This likely reduced the true difference in IOP GAT between cases and controls. Several studies have suggested an increase in CH in the first 6–12 months of PA use in treatment-naïve subjects.35–37 Tsikripis et al also suggested a small but significant increase in CCT with PA use.37 Conversely, Meda et al showed a reversible decrease in CH and CCT with PA treatment, but in subjects on long-term treatment.38 Pretreatment IOPcc, CH and CCT measurements were unavailable for both of our PA-treated subjects. As both were on long-term PA treatment, the findings of Meda et al may be more applicable. This may have resulted in a lower-than-expected CCT and CH, thus overestimating the difference between cases and controls.
Subject 8 was unable to comply with contact tonometry and pachymetry. Their IOPg measurement from the ORA was substituted when constructing the graphs. IOPg has been shown to correlate well with IOP GAT,12 15 so this substitution was not felt to have biased the mean IOP GAT difference between the groups.
Conclusion
In summary, the family members with a COL1A1 mutation had thinner corneas, lower corneal hysteresis and higher IOP than unaffected family members. Two of the cases had a known diagnosis of POAG and were on topical therapy and under regular clinical review. Our study supports the association made by Wallace et al and Mauri et al between COL1A1-related OI and POAG. Certainly, other genetic factors, in addition to the COL1A1 mutation, may coexist and predispose our subjects to POAG. Yet there is ample evidence to suggest that collagen abnormalities contribute in various ways to the development of POAG. Our findings of abnormal corneal properties in these subjects suggest that these non-invasive tests are a potential adjunct to diagnosis in patients with suspected OI, in the initial absence of definitive diagnostic genetic tests. More importantly, where a COL1A1-related OI diagnosis is confirmed, they are an invaluable screening tool for future glaucoma risk. Regular eye examinations should be carried out from a young age, and IOPcc as measured with the ORA may be a more accurate way to monitor IOP in the presence of abnormal corneal properties.
Data availability statement
All data relevant to the study are included in the article.
Ethics statements
Ethics approval
Ethics approval was not required as this is a case series wherein all investigations were done as part of appropriate clinical management and in the best interests of the subjects. Authors had full support and encouragement of the family and acquired consent forms from all subjects for genetic testing, and later for submission of article for publication.
Acknowledgments
Dr. Eoin Silke assisted with data collection.
References
Footnotes
Twitter @EmerDoolan
Contributors ED was responsible for data collection and composition of article. CO'B conducted literature search, edited the article and acts as guarantor.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement The patients reported on encouraged us to carry out the study, in terms of the examination of affected and unaffected members of the family as well as the genetic testing. They attended several meetings with the authors and discussed the results and their implications. They read the completed article prior to submission. They were not invited to contribute to the writing or editing of this document for readability or accuracy.
Provenance and peer review Not commissioned; externally peer reviewed.