Article Text

Abstract
Objective REFLECT is the first randomised, double-masked, placebo-controlled multicentre phase 3 clinical trial that evaluated the efficacy and safety of bilateral intravitreal (IVT) injection of lenadogene nolparvovec in subjects with Leber hereditary optic neuropathy carrying the m.11778G>A mutation.
Methods and analysis A total of 98 subjects were enrolled with vision loss of ≤12 months. The subjects were randomised to one of two treatment arms with all subjects receiving an intravitreal (IVT) injection of lenadogene nolparvovec in their first affected eye and the second-affected eye randomised to receive IVT of either lenadogene nolparvovec or placebo.
Results The majority of subjects were male with a mean duration of vision loss of 8.3 months. All but one subject experienced bilateral loss of vision at the time of injection. The mean best-corrected visual acuity of first-affected eyes was worse compared with second/not-yet-affected eyes. Analysis of retinal anatomical parameters showed increased thinning in the first-affected eyes when compared with the second/not-yet-affected eyes with both treatment arms showing significant changes compared with unaffected individuals.
Conclusion The REFLECT trial is the third and the largest phase 3 clinical study evaluating lenadogene nolparvovec in m.11778G>A Leber hereditary optic neuropathy (LHON) subjects. The observed demographics in REFLECT are consistent with previous reports in LHON subjects in the acute and dynamic phases of LHON disease. Combined with the visual function and anatomical parameters obtained in the previous RESCUE and REVERSE trials, REFLECT has provided a uniformly collected data set that should help direct future LHON clinical trials.
- Clinical Trial
- Pathology
- Vision
- Drugs
- Retina
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
The efficacy and safety of a unilateral intravitreal injection of lenadogene nolparvovec was evaluated in subjects with Leber hereditary optic neuropathy (LHON) carrying the m.11778G>A mutation in two previous phase 3 clinical studies.
WHAT THIS STUDY ADDS
The REFLECT trial is the only study to date evaluating the efficacy and safety of bilateral intravitreal injection of lenadogene nolparvovec in LHON subjects with the m.11778G>A mutation. REFLECT provides a large, homogeneous cross-sectional data set of visual function and retinal anatomic measurements collected during the acute and dynamic phases of the disease.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The results of REFLECT when combined with RESCUE and REVERSE provide important information on the inferred natural history of LHON that should help guide future research in subjects with the m.11778G>A mutation.
Introduction
Leber hereditary optic neuropathy (LHON), is a rare, maternally inherited mitochondrial genetic disease that typically causes severe bilateral visual loss from isolated optic neuropathies.1 Three primary point mutations in the mitochondrial DNA(mtDNA) at nucleotide positions m.3460G>A, m.11778G>A and m.14484T>C, occurring, respectively, in the ND1, ND4 and ND6 genes, have been identified as causative of LHON in approximately 90% of subjects. The primary mutations are necessary, but not sufficient, to cause vision loss, with approximately 20%–50% of male and 4%–10% of female carriers expressing the clinical disease, resulting in a male predominance of more than 80% in most pedigrees.1 Subjects are usually affected between the ages of 15 and 35 years, but symptomatic LHON has been reported in molecularly confirmed individuals as young as age 2 to as old as age 87.2–4 LHON classically manifests with acute to subacute, bilateral, painless central vision loss, with dyschromatopsia, central or cecocentral visual field defects, and often swollen-appearing hyperaemic optic discs.5
The REFLECT study (ClinicalTrials.gov NCT03293524) is a phase 3, international, multicentre, randomised, double-masked, placebo-controlled, clinical trial of ND4-LHON patients with vision loss ≤1 year in one or both eyes assessing the effect of bilateral intravitreal (IVT) injection of the gene therapy lenadogene nolparvovec. Lenadogene nolparvovec has been investigated previously in four clinical studies in 91 patients affected by vision loss from ND4-LHON: the REVEAL study, a phase 1/2a dose escalation open label study,6 and the RESCUE and REVERSE phase 3 randomised, sham-controlled pivotal studies.7–9 Patients who completed the RESCUE and REVERSE studies are currently followed in an ongoing extension study, RESTORE.10 The baseline characteristics of the 76 ND4-LHON patients from the RESCUE and REVERSE clinical trials presenting within 1 year of vision loss were recently published.11 This report describes the trial design of REFLECT and provides a cross-sectional analysis of the baseline characteristics of visual function and structural parameters in the 98 enrolled ND4-LHON subjects.
Methods
Study rationale
Lenadogene nolparvovec was investigated in four clinical studies (REVEAL, RESCUE, REVERSE, RESTORE) where it was exclusively administered as a unilateral IVT injection. However, LHON is a bilateral disease that typically affects both eyes of a patient in a sequential manner. The REFLECT study was therefore constructed to assess the efficacy and safety of bilateral IVT of lenadogene nolparvovec in MT-ND4 LHON subjects.
Study design
REFLECT (ClinicalTrials.gov NCT03293524) is a phase 3, international, multicentre, randomised, double-masked (for the primary analysis up to 1.5 years post-treatment), placebo-controlled, clinical trial conducted in 13 centres located across 7 countries (Belgium, France, Italy, Spain, Taiwan, United Kingdom (1 centre per country) and USA (7 centres)). Eligible subjects, LHON patients with vision loss ≤1 year in one or both eyes caused by the m.11778G>A mutation in the ND4 gene, were randomised into one of two treatment arms, with all patients receiving an IVT of lenadogene nolparvovec in their first affected eye and the second-affected eye randomised to receive IVT of either lenadogene nolparvovec or placebo (online supplemental figure 1). The primary efficacy analysis is the comparison of the change from baseline in best-corrected visual acuity (BCVA) reported at 1.5 years post-treatment between the second affected/not-yet-affected eyes receiving lenadogene nolparvovec and placebo.
Supplemental material
The study was conducted in accordance with the principles and requirements of the International Conference on Harmonisation Good Clinical Practice and adhered to the ethical principles outlined in the Declaration of Helsinki. An independent Data Safety Monitoring Board periodically reviews study data to ensure the continued safe conduct of the trial and protection of subjects.
Patient and public involvement
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Study population
The planned number of subjects to be included in the study was 90 overall (45 in each treatment arm) based on a treatment effect of 0.3 logarithm of the minimal angle of resolution (LogMAR) in the change from baseline to 1.5 years in BCVA, an SD of 0.50 and a power of 80% (alpha=0.05, two sided).
Inclusion and exclusion criteria
To be eligible for the REFLECT study, subjects needed to be 15 years of age or older with documented vision loss, to any extent, in at least one eye, documentation of the m.11778G>A mutation, duration of vision loss ≤365 days and a visual acuity of at least hand motion (HM) in each affected eye (online supplemental table 1). Patients using idebenone at screening were required to discontinue this treatment at least 7 days prior to inclusion. Exclusion criteria included contraindications to IVT, previous treatment with an investigational medicinal drug, ocular surgery within 90 days prior or IVT within 30 days prior, or history of recurrent uveitis or active ocular inflammation, and other causes of visual loss (eg, other optic neuropathies, macular disease) not attributable to LHON.
Assessments at baseline
Demographic characteristics were collected before treatment, along with visual function and anatomic parameters. Ophthalmologic assessments included BCVA, slit-lamp biomicroscopy, Goldmann applanation tonometry, funduscopy, contrast sensitivity (CS), automated perimetry, spectral-domain optical coherence tomography (SD-OCT) and colour fundus photography.
The BCVA was assessed using the Early Treatment Diabetic Retinopathy Study (ETDRS) charts at 1 or 4 m. Subjects who could not read at least three letters on a single line on the ETDRS chart at 1 m were tested for their ability to count the assessor’s fingers (CF), detect HM, and perceive light (light perception (LP)/no LP (NLP)). All BCVAs were expressed in LogMAR according to a standard conversion scale for on-chart values. Off-chart BCVA were assigned LogMAR values of+2.0 and +2.3, respectively, for CF and HM eyes, according to the validated Lange scale,12 and +4.0 and +4.5, respectively, for LP and NLP eyes.
CS was assessed with the Pelli-Robson Low Vision Contrast Sensitivity chart and expressed as a logarithm (LogCS).13 The Contrast Sensitivity score was defined as the last contrast triplet for which at least two of the three letters were read correctly. On-chart eyes were those able to read at least two letters of the first triplet on the chart and had a CS value expressed in logarithm (LogCS). Off-chart eyes were those unable to read at least two letters of the first triplet on the chart and were assigned a LogCS of 0 (worst possible score).
Standard, automated visual field assessment was obtained with an Humphrey Visual Field (HVF) Analyzer II (Carl Zeiss Meditec) using the 30–2 SITA Fast strategy. The HVF test was repeated if considered unreliable (ie, fixation losses ≥15%, false-positive errors ≥20% or false-negative errors ≥33%). The following parameters were collected: mean deviation (MD), pattern SD (PSD) and foveal threshold sensitivity (FT). If HVF foveal threshold was off then it was set to missing, and if the measured foveal threshold was <0, it was set to 0.
SD-OCT was performed using a Spectralis OCT (Heidelberg Engineering). Ganglion cell layer (GCL), temporal quadrant retinal nerve fibre layer (RNFL) thickness, papillomacular bundle RNFL thickness and average RNFL thickness were measured for the optic nerve and posterior pole as per standard protocols included in the Spectralis software. The OCT assessments were performed using triplicate scans of high quality (Q values >20). Borders of the retinal layers were manually adjusted when automated segmentation errors were detected.
BCVA, HVFs and SD-OCT exams were centrally reviewed, quality checked and graded by a central ophthalmology reading centre (William H. Annesley Jr. EyeBrain Centre (AEBC), Thomas Jefferson University). The AEBC was masked for all analyses.
Statistical analyses
Statistical analyses were carried out using SAS, software V.9.4 (SAS Institute). Summary statistics for continuous variables were described using N, mean, SD and range. The eyes with no vision loss were considered to have a duration of vision loss equal to 0. The predefined baseline for visual function (BCVA, CS, HVF) and anatomic parameters (OCT) was the last available assessment before treatment; for anatomic metrics, the average of values measured at screening and inclusion visits was also calculated.
Regressions between baseline parameters were performed with an analysis of covariance (ANCOVA) model which includes terms treatment as a fixed effect and with repeated values for eye. The partial correlation coefficient was performed to quantify the impact of one variable on a second variable. From the ANCOVA model the correlation is calculated as:
 where
where  for m subjects.
for m subjects.  and
and  are the model estimates and variances.14
are the model estimates and variances.14
Results
Disposition
A total of 108 patients were screened and 98 were included, randomised, and treated from March 2018 to June 2019.
Demographic characteristics
Of the 98 included ND4-LHON patients, 78 (79.6%) were male and the average age at disease onset was 31.5 years (range 14–73 years) (online supplemental table 2). Ten subjects (10.2%) were between 15 and 18 years of age, and 5 subjects (5.1%) were at least 60 years old. There were 15 subjects (15.3%) from Asia and the remaining subjects were from either the USA (56 subjects, 57.1%) or Europe (27 subjects, 27.5%). Half of patients reported alcohol consumption and approximately one in five subjects were current smokers. Prior idebenone treatment was reported by 14 subjects (14.3%), and all discontinued the treatment at least 7 days prior to inclusion, consistent with protocol requirements.
Disease characteristics
Duration of vision loss at baseline averaged 8.3 months (range of 1.7–11.9), consistent with inclusion criteria requiring vision loss ≤1 year. Among the 98 patients, one subject had unilateral disease (only one eye affected) at the time of treatment with the fellow eye having a BCVA of 20/20 at baseline (online supplemental table 3).
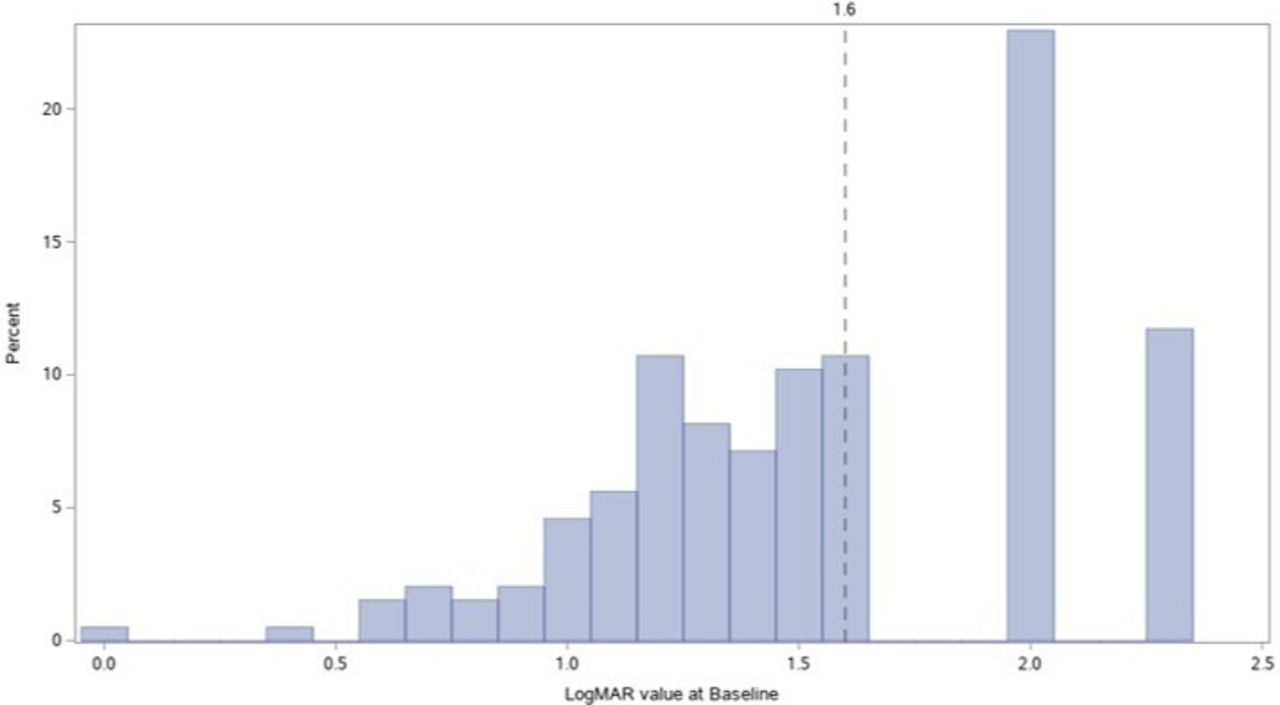
The mean BCVA of the first-affected eyes was worse than that of the second affected eyes (1.64 LogMAR vs 1.47 LogMAR) (online supplemental table 2). More than one third of eyes were off-chart at baseline (68 eyes; 34.7%) and 25 subjects had both eyes off-chart. The distribution of BCVAs for all eyes is provided in figure 1.

Histogram of best-corrected visual acuity values in logMAR at baseline (n=196 eyes). logMAR, logarithm of the minimal angle of resolution.
A total of 89 eyes (45.4%) were off-chart for CS at enrolment (these eyes could not correctly read 2 letters at the maximum contrast possible on the Pelli-Robson chart). The mean Log CS was 0.37, with a range from 0 to 1.35 LogCS; off-chart eyes were scored 0 for LogCS (online supplemental table 2).
The mean (SD) MD and PSD for HVF 30–2 testing in all eyes were −22.31 (10.3) dB and 7.02 (3.5) dB, respectively.
LogMAR and HVF MD showed no significant correlation with tobacco and alcohol consumption (online supplemental tables 4 and 5).
Retinal anatomy analyses revealed that the average values of retinal layers of interest were consistently thinner for the first affected eyes when compared with the second affected eyes, with both differing significantly from population norms (online supplemental table 2).
Association between baseline parameters
At baseline, a correlation was observed between the GCL macular volume and the BCVA: the less affected BCVAs were associated with larger GCL macular volumes (correlation coefficient; r=−0.668) (figure 2). A similar relationship was observed between the average RNFL thickness and the BCVA (r=−0.637) (figure 2).
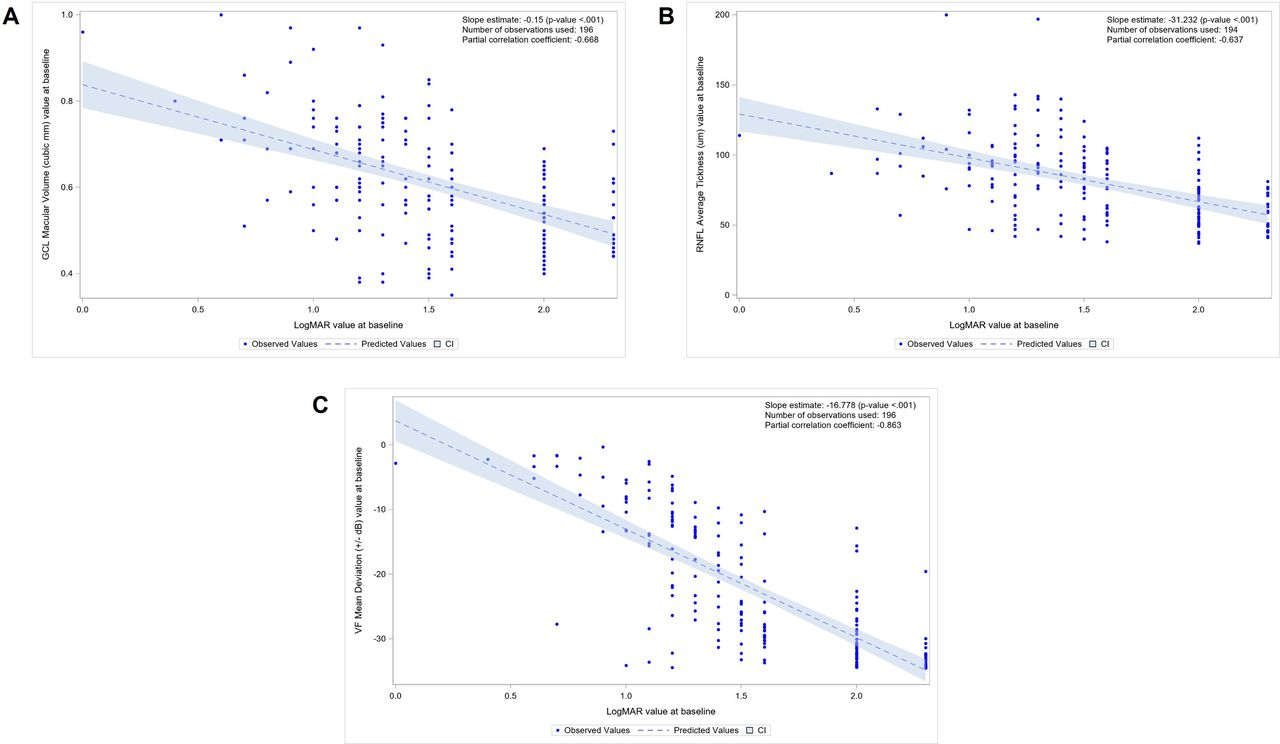
(A) Regression analysis between ganglion cell layer (GCL) macular volume and best-corrected visual acuity (BCVA) in logMAR at baseline (n=196 eyes). (B) Regression analysis between average retinal nerve fibre layer (RNFL) thickness and BCVA in logMAR at baseline (n=196 eyes). (C) Regression analysis between humphrey visual field (HVF) mean deviation and BCVA in logMAR at baseline (n=196 eyes). logMAR, logarithm of the minimal angle of resolution.
As observed for the OCT values, the baseline visual field measurements, assessed by the HVF MD, were related to the baseline BCVA: the less affected BCVAs were correlated with less severe MD (r=−0.863) (figure 2).
Similarly, better LogCS values were associated with lesser reductions in GCL macular volume (r=0.675), average RNFL thickness (r=0.671) and HVF MD (r=0.892) (figure 3).
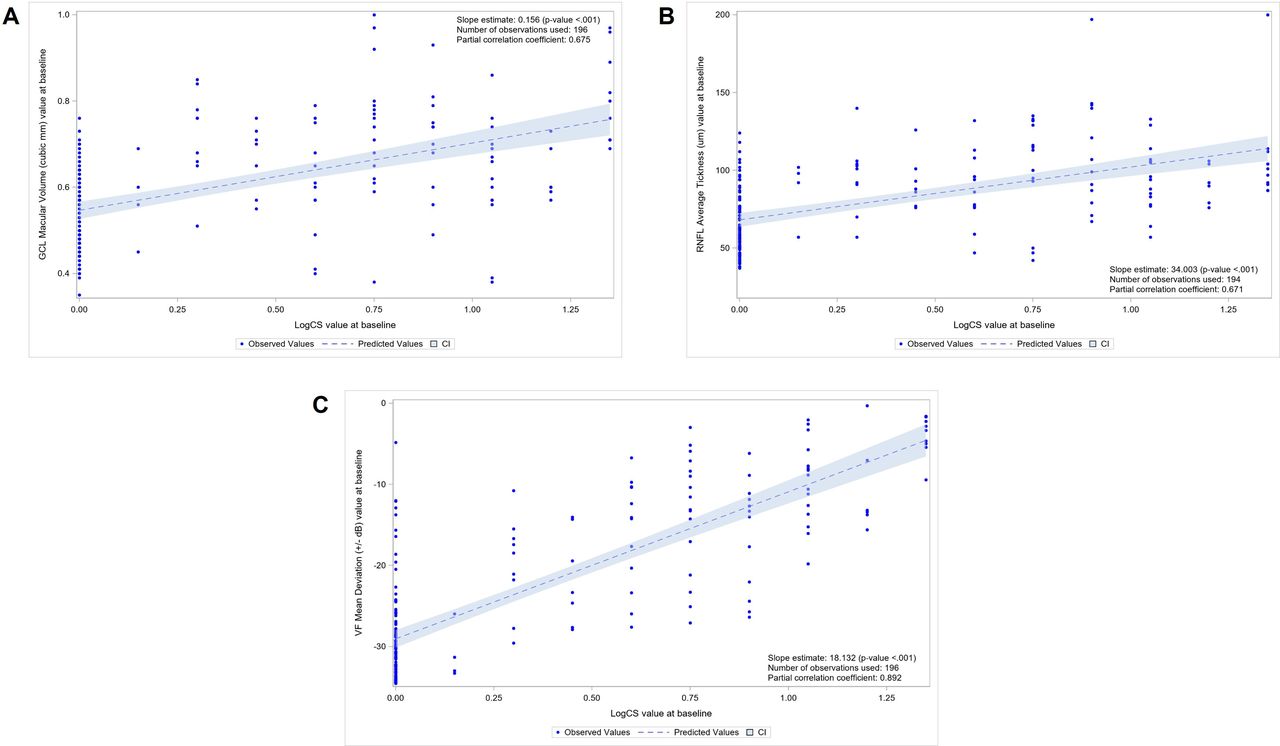
(A) Regression analysis between ganglion cell layer (GCL) macular volume and logarithm of contrast sensitivity (LogCS) at baseline (n=196 eyes). (B) Regression analysis between average retinal nerve fibre layer (RNFL) thickness and LogCS at baseline (n=196 eyes). (C) Regression analysis between humphrey visual field (HVF) mean deviation and LogCS at baseline (n=196 eyes).
There was also a correlation between LogMAR and LogCS values at baseline (r=−0.878) (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Regression analysis between logarithm of contrast sensitivity (LogCS) and best-corrected visual acuity (BCVA) in logMAR at baseline (n=196 eyes). LogMAR, logarithm of the minimal angle of resolution.
Discussion
We present a large, homogeneous dataset of visual function and retinal anatomic measurements collected during the first year following visual loss in LHON subjects carrying the m.11778G>A mutation in the ND4 gene. These results provide detailed information on the first two clinical stages of the disease (subacute (<6 months from onset) and dynamic (6–12 months)) prior to onset of the chronic stage.15 To our knowledge, the REFLECT population is the largest ND4-LHON cohort studied in a randomised, double masked, controlled study, providing meaningful information on the inferred natural history of disease, using cross-sectional analysis on 98 patients (196 eyes). The REFLECT demographic characteristics are aligned with the typical ND4-LHON population, as described in the literature.16–18 Our results confirm the male predominance (80%) and a preponderance of young adults (median age: 28.0 years), recognising that our study only included patients at least 15 years of age. Visual impairment of the second eye occurred quite rapidly, on average approximately 2 months after the first eye. Consistently, the first affected eyes, with a longer duration of vision loss at baseline, had worse visual function and anatomic parameters than the more recently second affected eyes. The difference in HVF parameters was less striking between the first and second affected eyes but was still present. There was no discernible influence of smoking or alcohol consumption on baseline BCVA or HVF MD.
There was a correlation between anatomic parameters and BCVA at baseline using the cross-sectional analyses over the 1-year postvision loss . In addition, the correlation was even stronger between visual function parameters: HVF and BCVA . The same magnitude of correlation of OCT parameters and HVF MD with LogCS was observed, with again a stronger correlation with HVF MD. Furthermore, a correlation between BCVA and LogCS was present, as both parameters showed a comparable magnitude of correlation with OCT parameters and HVF MD .
RESCUE and REVERSE (ClinicalTrial.gov NCT02652767and NCT026527080)7 8 were two randomised, double masked, sham-controlled, multicentre, international clinical trials assessing the benefit of a single IVT of lenadogene nolparvovec in 76 patients. In RESCUE, one or both eyes could be affected by vision loss provided the duration of vision loss was ≤6 months in the first (or only) affected eye at screening. In REVERSE, both eyes had to be affected by vision loss for 6–12 months at time of screening. To be included in either study, LHON subjects had to harbour the m.11778G>A mutation, be at least 15 years old at enrolment, and have vision of CF or better in both eyes. Baseline data analyses of these two studies were recently published.11 The REFLECT demographic characteristics are comparable to the REVERSE and RESCUE populations’ characteristics (table 1). The mean time for inclusion and treatment for RESCUE, REFLECT and REVERSE subjects was 112, 223 and 271 days, respectively, in alignment with the inclusion criteria of each trial. As expected, the difference in duration of vision loss was associated with a consistent gradient of impairment of visual function and anatomic parameters at baseline across the three clinical studies populations (worse in REVERSE, intermediate in REFLECT, better in RESCUE) (table 1). In addition, the one REFLECT subject with unilateral vision loss at baseline had similar structural and functional characteristics to four patients with unilateral vision loss in RESCUE (online supplemental table 6).
Comparison of baseline characteristics of visual function and retinal anatomy in the rescue, reflect and reverse studies
The limitations of our analyses are related to the cross-sectional nature of the data, as subjects naive to LHON treatment were subsequently administered gene therapy and not followed longitudinally as untreated individuals. However, the data assembled from the REFLECT study provide a valuable landscape of the clinical characteristics of LHON patients harbouring the m.11778G>A mutation in MT-ND4 within 1 year of the onset of vision loss. In addition, when combined with the baseline data from the RESCUE and REVERSE trials, these data improve our understanding of the subacute and dynamic phases of vision loss from ND4-LHON and provide uniformly collected anatomic and functional information to guide future interventions.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Belgium: Universitair Ziekenhuis Gent (2018/0074)France: Comité de Protection des Personnes Nord-Ouest II (2018/15)Italy: Comitato Etico Interaziendale Bologna-Imola - CEBI (88929/13/07/2018)Spain: CEIC Hospital Universitario Ramon y Cajal (18.01.1161-GHM)Taiwan: Taipei Veterans General Hospital, Institutional Review Board (2018-03-002A)UK: NRES Committee - London — West London & GTAC (18/LO/0521)USA: Wills Eye Hospital,IRB (15.1118); Emory University-EC (IRB00100950); UCLA Medical Center IRB (18-000742); WIRB (20180153); Human Studies Committee (18-051H); Mount Sinai School of Medicine (IBC-2018-041); Vanderbuilt IRB (180255). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We thank Julie Salzmann and Anne-Coline Laurent for their medical writing support for this manuscript. Patrick Yu-Wai-Man is supported by an Advanced Fellowship Award (NIHR301696) from the UK National Institute of Health Research (NIHR). Patrick Yu-Wai-Man also receives funding from Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity (GR001376), the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the International Foundation for Optic Nerve Disease (IFOND), the NIHR as part of the Rare Diseases Translational Research Collaboration, the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014), and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Collaborators LHON REFLECT Study Group:Amore Giulia, Anand Shweta, Banik Rudrani, Barboni Piero, Biousse Valérie, Boston Hayley, Burale Asma, Carbonelli Michele, Carelli Valerio, Chen Celia, Cheng Hui-Chen, Cho Steve, Chwalisz Bart K, Contin Manuela, D’Agati Pietro, DeBusk Adam A, De Zaeytijd Julie, Dobbs Jannah, Donahue Sean P, DuBois Lindreth, Esposti Simona, Fernandes Filho Alcides, Fortin Elizabeth, Gangaputra Sapna, Gibbs Deborah, Girmens Jean François, Hage Rabih, Haller Julia A, Heilweil Gad, Hubbard III George Baker, Hwang Jeong-Min, Jaumendreu Urquijo Laia, Jurkute Neringa, Karanjia Rustum, Khemliche Wahiba, La Morgia Chiara, Leroy Bart P, Massini Maria, Mathias Marc, Memon Muhammad A, Mohamed Susan, Moster Mark L, Muñoz Negrete Francisco J, Newman Nancy J, O’Keefe Ghazala, Patel Shriji, Pecen Paula, Peragallo Jason H, Plaine Lise, Preston Mary, Rebolleda Fernández Gema, Romagnoli Martina, Sadun Alfredo A, Sahel José A, SantaMaria Melissa, Sergott Robert C, Subramanian Prem S, Sun Chuanbin, Tai Katy, Tollis Heather, Tsui Irena, Tucker William R, Vignal-Clermont Catherine, Wang An-Guor, Wilkins Saige, and Yu-Wai-Man Patrick.
Contributors Guarantor: PSS. Conception and design: NJN, PY-W-M, VC, VB, CV-C, RCS and J-AS. Data collection: The LHON REFLECT Study Group and FB, MR, EC and MT. Analysis and interpretation: NJN, PY-W-M, VC, VB, CV-C, RCS, PSS, MM, SD, FB, MR, EC, MT and J-AS.
Funding This study was funded by Gensight Biologics.
Competing interests PSS is a consultant for GenSight Biologics, Horizon Therapeutics, Invex Therapeutics, Viridian Therapeutics, and Kriya Therapeutics and has received research support from Santhera Pharmaceuticals, GenSight Biologics, and Horizon Therapeutics; and is a medical legal consultant. NJN is a consultant for GenSight Biologics, Santhera Pharmaceuticals, and Stealth BioTherapeutics; has received research support from GenSight Biologics and Santhera Pharmaceuticals; served on the Data Safety Monitoring Board for Quark NAION study; and is a medical legal consultant. MM is a consultant for GenSight Biologics and has received research support from GenSight. PY-W-M is a consultant for GenSight Biologics and Stealth BioTherapeutics and has received research support from GenSight Biologics and Santhera Pharmaceuticals. SD has been a fee-for-service consultant for GenSight Biologics. BPL is a consultant for GenSight Biologics, 4DMT, AAVantgardeBio, Akouos, Asthena Therapeutics, Bayer, Biogen, GenSight Biologics, IVERIC Bio, MeiraGTx-Jansen Pharmaceuticals, LookoutGTx, Novartis, Opus Genetics, Oxurion, ProQR Therapeutics, Santen, Spark Therapeutics, REGENXBIO, Vedere Bio, ViGeneron. VC is a consultant for GenSight Biologics, Santhera Pharmaceuticals, and Stealth BioTherapeutics and has received research support from Santhera Pharmaceuticals and Stealth BioTherapeutics. VB is a consultant for GenSight Biologics. CVC is a consultant for Santhera Pharmaceuticals and GenSight Biologics. RCS is a consultant for GenSight Biologics. AAS is a consultant for Stealth BioTherapeutics. RB is a consultant for Horizon Therapeutics, Healthy Directions and Guardion Health Sciences and has received research support from Santhera Pharmaceuticals, Regenera and Quark Pharmaceuticals. EC, Michel Roux and Magali Taiel are employed by GenSight Biologics, the sponsor of these studies. JAS is the cofounder and shareholder of GenSight Biologics and the patent coauthor on allotopic transport.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.