Article Text

Abstract
Objective Paediatric (childhood or congenital) cataract is an opacification of the normally clear lens of the eye and has a genetic basis in at least 18% of cases in Australia. This study aimed to replicate clinical gene screening to identify variants likely to be causative of disease in an Australian patient cohort.
Methods and analysis Sixty-three reported isolated cataract genes were screened for rare coding variants in 37 Australian families using genome sequencing.
Results Disease-causing variants were confirmed in eight families with variant classification as ‘likely pathogenic’. This included novel variants PITX3 p.(Ter303LeuextTer100), BFSP1 p.(Glu375GlyfsTer2), and GJA8 p.(Pro189Ser), as well as, previously described variants identified in genes GJA3, GJA8, CRYAA, BFSP1, PITX3, COL4A1 and HSF4. Additionally, eight variants of uncertain significance with evidence towards pathogenicity were identified in genes: GJA3, GJA8, LEMD2, PRX, CRYBB1, BFSP2, and MIP.
Conclusion These findings expand the genotype–phenotype correlations of both pathogenic and benign variation in cataract-associated genes. They further emphasise the need to develop additional evidence such as functional assays and variant classification criteria specific to paediatric cataract genes to improve interpretation of variants and molecular diagnosis in patients.
- Genetics
- Child health (paediatrics)
- Lens and zonules
- Experimental & laboratory
Data availability statement
Data are available on reasonable request. All variants reported in this study have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); ClinVar accession numbers SCV001573165-SCV001573189. Additional data can be made available on request if within the ethical bounds of this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Paediatric (congenital) cataract is a genetically and phenotypically heterogeneous disease. Genomic testing of children with cataract is increasingly being used to refine the diagnosis, determine prognosis and guide genetic counselling.
WHAT THIS STUDY ADDS
Three novel disease-causing variants, in genes PITX3, BFSP1 and GJA8, expand the genotype–phenotype spectrum of paediatric cataract and several previously described variants re-enforce their pathogenic classifications. Eight families had ‘variants of uncertain significance’ that could be reclassified as likely pathogenic with additional evidence.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Stringent classification of variants is required for clinical genetic testing and highlights that many variants do not have sufficient evidence for a clinically actionable classification, even in well-characterised disease-causing genes.
Introduction
Paediatric cataract (childhood or congenital cataract), a clouding of the crystalline lens of the eye during childhood, is a heritable condition in at least 18% of cases in Australia.1 The total number of genes associated with a cataract phenotype is in excess of 200.2 However, a core set of approximately 40 well-established genes are known to cause isolated paediatric cataract that include: crystallin genes (CRYAA, CYRAB, CRYBA1, CRYBA2, CRYBA4, CRYBB1, CRYBB2, CRYBB3, CRYGA, CRYGB, CRYGC, CRYGD and CRYGS), genes encoding membrane structural proteins (GJA3, GJA8, MIP and LIM2) and cytoskeletal proteins (VIM, BFSP1 and BFSP2), transcription factor genes (HSF4, PITX3, PAX6, FOXE3 and MAF) and genes for signalling molecules such as EPHA2. Other genes, such as NHS, FTL, AGK, MIR184, GCNT2 and GALK1, are also routinely assessed and are associated with either isolated paediatric cataract or paediatric cataract as a characteristic phenotype as part of a syndrome. The success of genetic screens for familial paediatric cataract has varied greatly with reported solve rates between 25% and 77%.3 4 Cohort, gene panel selection, sequencing methodology and the stringent use of variant classification criteria will have contributed to the varied successes. Despite this, routine gene screening and variant reporting in the research setting plays a very important role in expanding the known genetic and phenotypic spectrum of this condition, particularly regarding novel and less-established cataract-associated genes. Variant reporting of both negative and positive findings improves the collective understanding of these genes, their products and the mechanisms that cause cataracts, and ultimately improves clinical diagnosis and outcomes for patients.
This study aimed to investigate a panel of 63 isolated cataract-associated genes in 37 Australian families. Disease-causing likely pathogenic variants in genes GJA3, GJA8, CRYAA, BFSP1, PITX3, COL4A1 and HSF4 were identified in eight families. Variants of uncertain significance (VUS) were identified in a range of well-established and other cataract genes that may be disease-causing but require additional evidence of pathogenicity following current scoring with the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) guidelines.
Materials and methods
All affected participants were diagnosed with inherited paediatric cataract, based on the observed cataract phenotype and a reported family history, by the examining ophthalmologist and genetic counsellors, respectively. Relatives were examined on recruitment to the study or following diagnosis during routine examination anytime thereafter. DNA was extracted from whole blood using a QiaAmp DNA blood Maxi Kit (Qiagen), buccal mucosa swabs using the PureGene DNA Isolation Kit (Gentra Systems) or saliva using Oragene DNA saliva collection kits (DNA Genotek, Ontario, Canada). Clinically actionable variants were returned to patients as research findings through our genetic counsellors, who facilitate subsequent nationally accredited genetic testing in Australia and appropriate counselling.
Genome sequencing was performed on the DNA of an affected individual (proband) from each of 37 families with European ancestry. Sequencing was either 150 bp paired-end sequencing on a HiSeq X Ten platform (30× coverage, Illumina) with an Illumina TrueSeq Nano Library Prep (V.2.5) at the Kinghorn Centre for Clinical Genomics (Sydney, Australia) or 250 bp paired-end sequencing on a NovaSeq 6000 platform (30× coverage, Illumina) with an Illumina Nextra DNA Flex library preparation at the Ramaciotti Centre for Genomics (Sydney, Australia). Variant calling was performed using the bcbio-next-gen pipeline (https://doi.org/10.5281/zenodo.3564938) with the BWA-MEM algorithm5 for read alignment to human reference genome hg19 and variant calling with GATK6 according to best practice guidelines. Variant annotation was performed using ANNOVAR.7 MultiQC8 reporting was used to assess sample quality and average target coverage, and read depth at variant sites exceeded 30 for most samples (online supplemental table S1). Five samples with lower target coverage and read depth were retained, although interpreted with caution.
Supplemental material
Sixty-three genes were selected for assessment (online supplemental table S2) based on previous well-established congenital cataract genes, research-reported candidate genes or genes that are otherwise associated with syndromic conditions or other ocular phenotypes that have reports of isolated congenital cataracts or cataracts as an early presenting feature. The gene list is comparable to the panels used by accredited genetic testing laboratories for non-syndromic paediatric cataract. Variants within those genomic regions were filtered to functional ‘exonic’ or ‘splicing’ variants, with an MAF ≤0.00022 in gnomADv2.1.19 pop_max (highest population frequency) to match the reported Australian disease frequency of 2.2 per 10 000 live births.1 Variants were prioritised for further analysis if they had a CADD PHRED score ≥10 or, for synonymous and non-coding RNA variants, a CADD PHRED score ≥15. Variant validation and cosegregation was performed with primers designed using NCBI primer blast10 (online supplemental table S3). PCR was performed using MyTaq HS DNA polymerase (Bioline) prior to Sanger sequencing with either BrightDye Terminator (MCLAB) or BrilliantDye Terminator (Nimagen) Cycle sequencing kits and sequenced using an ABI 3500 Genetic Analyzer (Life Technologies), all according to manufacturer’s instructions.
Predictive analysis of protein folding between wild-type and variant protein sequences was performed using the PredictProtein folding prediction tool (https://predictprotein.org).11 12 HOPE protein structure analysis13 was used to further assess the deleteriousness of missense variants on protein function. The mFold tool14 (http://www.unafold.org/) was used for comparing microRNA folding between wild-type and variant sequences. Validated variants were interpreted using the ACMG-AMP guidelines15 via InterVar16 and manually adjusted as appropriate. Cosegregation considered based on informative meioses17 and PP5 and BP6 criteria were excluded from use.18 All variants reported in this study have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); ClinVar accession numbers SCV001573165-SCV001573189. Evidence for pathogenicity according to the ACMG-AMP guidelines is shown in online supplemental table S4).
Results
Sixty-three paediatric cataract genes were screened in probands from 37 Australian families with inherited paediatric cataract. This cohort contained 13 probands/families that remained unsolved following a screen of 51 genes (families indicated in online supplemental table S1).19 The other 24 probands/families were either unsolved following analysis of the NHS,20 EPHA221 and crystallin genes22 or have not previously been investigated.
Likely pathogenic variants
Eight probands/families had variants classified as likely pathogenic (8 of 37, 22%), including of three novel and five previously described variants (table 1).
Rare coding variants detected in probands with paediatric cataract
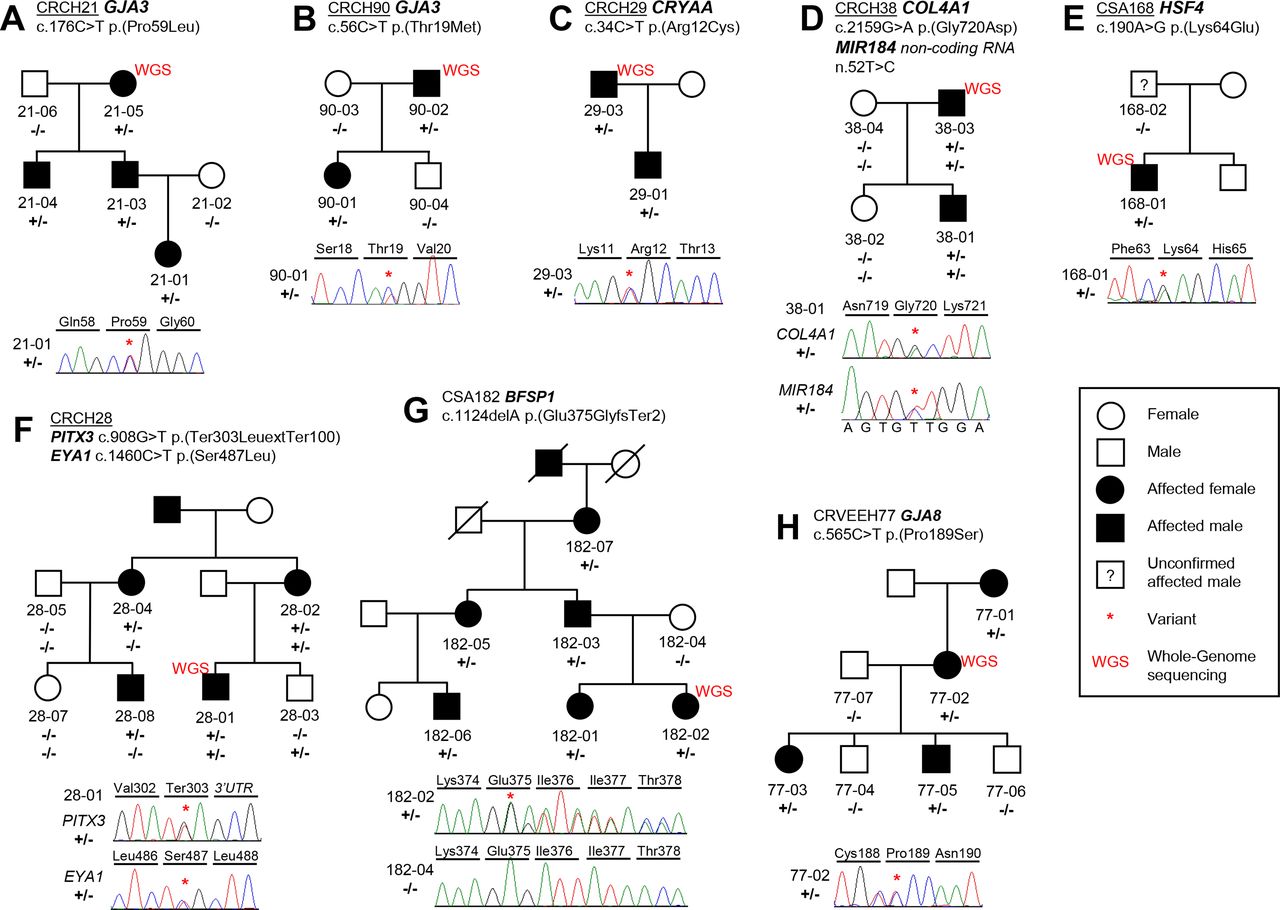
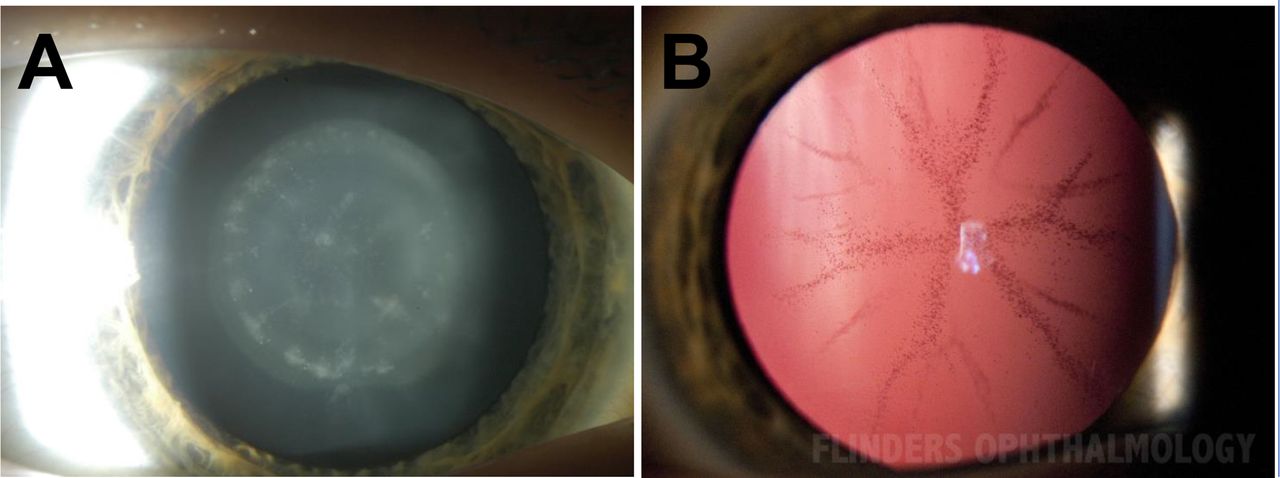
Two previously described variants were identified in the GJA3 gene (p.Pro59Leu and p.Thr19Met) in families CRCH21 and CRCH90 (figure 1A–B). A commonly reported pathogenic CRYAA (p.Arg12Cys) change was identified in family CRCH29 (figure 1C). A known COL4A1 p.(Gly720Asp) change was determined to be disease-causing in family CRCH38 (figure 1D). Additionally, in family CRCH38, a MIR184 n.52T>C change was also observed but deemed benign with no harmful predicted change to secondary structure of the microRNA molecule (online supplemental figure S1). A p.(Lys64Glu) HSF4 change was identified in family CSA168 (figure 1E) and is located in the highly conserved DNA binding domain of the protein. This HSF4 variant has previously been reported as pathogenic23 in a family with lamellar paediatric cataract, with a comparable phenotype observed here in CSA168-01 (figure 2A).

Families with likely pathogenic variants in isolated paediatric cataract causing genes. (A) Family CRCH21 with a segregating previously described GJA3 p.(Pro59Leu) variant. (B) Family CRCH90 with a previously described GJA3 p.(Thr19Met) variant. (C) Family CRCH29 with a previously described CRYAA p.(Arg12Cys) variant. (D) Family CRCH38 with previously described COL4A1 p.(Gly720Asp) variant and MIR184 n.52T>C variant of uncertain significance. (E) Family CSA168 with previously described HSF4 p.(Lys64Glu) variant in the proband. (F) Family CRCH28 with a novel segregating PITX3 p.(Ter303LeuextTer100) variant and non-segregating EYA1 p.(Ser487Leu) variant. (G) Family CSA182 with a novel BFSP1 c.1124delA frameshift variant. (H) Family CRVEEH77 with a novel segregating GJA8 p.(Pro189Ser) variant altering an amino acid that has been previously associated with paediatric cataract.

Cataract phenotypes. (A) Lamellar cataract observed in CSA168-01 via slit-lamp photography using direct illumination. (B) Transillumination of CSA182-06, displaying the posterior sutural cataract phenotype with a pulverulent appearance was consistently observed in all affected individuals in the family.
A novel stop loss PITX3 variant, in family CRCH28 (figure 1F), was predicted to extend the normal 302-residue long protein by an additional 100 amino acids, when assessed using the NCBI ORF finder (https://www.ncbi.nlm.nih.gov/orffinder/). Comparative protein folding prediction indicated small structural changes within the DNA binding domain and the formation of beta-strands with potential DNA-binding and protein-binding affinity in the additional 100 amino acid protein extension (online supplemental figure S2). A benign EYA1 variant was also observed but failed to cosegregate with the disease in the family. Affected individuals were diagnosed between 9 and 21 years of age with posterior subcapsular cataracts (table 1) and had no other reportable ocular features. This cataract phenotype is consistent with the posterior polar or posterior subcapsular opacifications reported in PITX3 variants to date, with or without additional anterior segment mesenchymal dysgenesis features.2
A novel BFSP1 c.1124delA p.(Glu375GlyfsTer2) frameshift variant segregated in an autosomal dominant manner in family CSA182 (figure 1G). A posterior sutural cataract phenotype with a pulverulent appearance (figure 2B) was consistently observed across all affected individuals in the family. Sutural, often pulverulent-like, cataracts are the most frequently reported phenotype with BFSP2 variants while the range of phenotypes observed for BFSP1 variants is wider and includes nuclear, lamellar and cortical cataracts.2
In family CRVEEH77, a novel p.(Pro189Ser) GJA8 variant was identified. Previous reports of variants c.565C>G p.(Pro189Ala) and c.566C>T p.(Pro189Leu) have been made at this location in patients with isolated paediatric cataracts, all with nuclear or nuclear inclusive phenotypes.24 25 Additionally, the same residue change to the equivalent conserved amino acid in the GJA3 protein, p.(Pro187Ser), has been reported as disease-causing.26
VUS with evidence towards pathogenicity
Eight probands/families had VUS with evidence towards pathogenicity (22%, table 1). Six of these reside in well-established isolated cataract genes GJA3, GJA8, CRYBB1, BFSP2 and MIP. With additional evidence of pathogenicity, such as additional meiosis demonstrating segregation or robust functional evaluation, these variants would likely be upgraded in classification to pathogenic variants.
Three variants were identified in connexin genes GJA8 and GJA3. A GJA8 p.(Gly22Ser) change, observed in family CRCH137, has been previously reported25 27 28 and is one meiosis short of reclassification as likely pathogenic (table 1, figure 3A). In family CTAS71, the novel p.(Lys131del) change (table 1, figure 3B) is located centrally in the GJA8 protein’s cytoplasmic loop which is a less conserved protein region in general. A GJA3 c.43C>A p.(Gln15Lys) variant was observed in singleton CSA192 (table 1, figure 3C). This glutamine residue is in a very highly conserved region of the GJA3 protein, with only asparagine and arginine alternatively observed at this position in the softshell turtle and tetraodon fish, respectively. The CTAS71 and CSA192 variants are likely de novo but require parental analysis.

{kind=link}
{kind=link}
{kind=link}
Families with variants of uncertain significance that have evidence towards pathogenicity. Families with gap junction variants include: (A) GJA8 p.(Gly22Ser) in family CRCH137, (B) GJA8 p.(Lys131del) in family CTAS71 shown in ‘R’ for sequencing with reverse primer, and (C) GJA3 p.(Gln15Lys) in family CSA192. Five families were observed to have variants of uncertain significance in other cataract associated genes. (D) family CRVEEH79 with a start loss variant in the LEMD2 gene. (E) Family CSA93 with a segregating PRX p.(Arg129His) change. (F) Family CTAS34 with a segregating CRYBB1 p.(Ile94Asn) variant and MIP variant was also observed but also present in an unaffected individual. (G) family CQLD130 with a BFSP2 p.(Arg89Trp) variant that is also observed in unaffected CQLD130-04 and is acting with possible reduced penetrance. (H) In family CRCH4 a MIP p.(Arg113Gln) variant was observed in the two affected individuals and obligate heterozygote CRCH4-03, as well as, unaffected siblings CRCH4-06 and CRCH4-08.
In family CRVEEH79, affected individuals were heterozygous for a start-loss variant in the LEMD2 gene (table 1, figure 3D). Both individuals have mild blue dot cataracts that have not yet required surgery. The LEMD2 c.1A>G variant was predicted to use an alternative methionine at position 233 in the native protein sequence, resulting in the loss of the conserved LEM domain, lamin A/C complex interacting region and one of two transmembrane domains. Alternatively, a methionine in a different reading frame closer to the 5’-untranslated region could be recruited and produce an 86 amino acid long protein or transcript likely to be subject to nonsense-mediated decay, which would result in a null allele.
The PRX c.386G>A variant observed to cosegregate in family CSA93 (table 1, figure 3E) causes a p.(Arg129His) change in the S-periaxin encoding NM_020956.2 transcript only, and is located in the intron of the transcript encoding the L-periaxin isoform c.381+5G>A (NM_181882.3). The variant causes the non-conserved arginine to be replaced with a smaller but still positively charged histidine residue at the C-terminal end of the translated protein.
A CRYBB1 p.(Ile94Asn) change was identified in CTAS34 that likely accounts for their disease (table 1, figure 3F). Three residue types are observed across species at this site (Ile, Leu, Val), all of which are nonpolar compared with the polar asparagine reported in this family. Located in the first of four Greek Key motifs, the incorporation of a polar residue at this site was predicted to disrupt the hydrophobic interactions in the core of the protein.13 Cataract-causing variants have been reported at adjacent residues p.Ser9329 and p.Val9630 indicating a region of functional importance. Additional segregation evidence from other known affected family members would be highly valuable and would assist in upgrading the classification of this variant. The MIP p.(Val164Ile) variant in the same family was also classified as a VUS, however, based on its higher population allele frequency, non-damaging in silico predictions and presence in an unaffected individual it is unlikely to be disease-causing.
In family CQLD130, the BFSP2 p.(Arg89Trp) variant was observed in four affected individuals and a child who was unaffected as the 10 years old at last examination, but not yet old enough to be confirmed as unaffected for a childhood onset disease (table 1, figure 3G). This residue change occurs within an evolutionary constrained block of the phakinin protein’s N-terminus head region (amino acid 1–114) prior to the main α-helical rod that forms the majority of the 312 amino-acid-long structure. At this site arginine is observed in most species; however, tryptophan has been observed in some species. Changes to this BFSP2 VUS classification will depend on future surveillance of CQLD130-04 for cataract development or additional reports in unrelated cataract patients.
In family CRCH4, the identified MIP p.(Arg113Gln) variant was assessed as being potentially disease-causing (table 1, figure 3H). The p.Arg113 residue is highly conserved across species, with positively charged residues in this extracellular domain known to be functionally important for AQP0 in cell–cell adhesion.31 Reduced penetrance is observed in this family with obligate heterozygote CRCH4-03 showing no clinically significant opacities. Affected individual CRCH4-01 was diagnosed at birth and received surgery within a month, whereas CRCH4-05 was diagnosed at 2 years of age and did not require surgery until 38 years of age. The two other variant carriers, CRCH4-06 and CRCH4-08, were 7 and 4.5 years of age at last examination, respectively, at which time both still had clear lenses. This family is being screened regularly to assess for cataract development in unaffected individuals.
VUS that are unlikely to be disease-causing
Five families had identified variants classed as VUS unlikely to be causing disease (14%, table 1, online supplemental figure S3). In family CSA158, a CYP51A1 p.(Ala94Thr) variant was the only variant observed to fully cosegregate with disease (online supplemental figure S3A) but is inconsistent with the recessive inheritance patterns that have been previously reported with variants in this gene and other cataract-associated genes involved in the cholesterol biosynthesis pathway. The primary evidence against pathogenicity in the remaining four families was poor cosegregation of the variant with disease in the context of reported inheritance patterns for the gene, population allele frequency and in silico predictions (online supplemental figure S3B-E).
Discussion
A comprehensive selection of 63 isolated cataract-associated genes in 37 Australian families were investigated. Disease-causing likely pathogenic variants were identified in eight families in genes GJA3, GJA8, CRYAA, BFSP1, PITX3, COL4A1 and HSF4. An additional eight families were identified to have VUS with evidence towards pathogenicity. The solved rate for this cohort resides between 22% and 43%, with many of the identified VUS likely to be disease-causing with the acquisition of additional evidence. With a subset of patients previously cleared of variants in cataract genes there was a reduced likelihood of achieving a solved rate comparable to screening a previously unstudied population. Despite this, these rates are not dissimilar to the 42% likely disease-causing in the previous study of our repository19 and the recent 44.4% molecular diagnostic rate reported for clinical congenital cataract screening in the UK.32
Variants in connexin and crystallin genes again accounted for approximately half of disease-causing variants in Australian cohorts.4 19 These gene products play critical and well-characterised roles in maintaining lens homeostasis and creating a high protein content that aids in achieving lens transparency.
Classification of the COL4A1 p.(Gly720Asp) change as likely pathogenic, in family CRCH38, was greatly assisted by its previous observation in a family with a congenital cataract phenotype.33 34 All individuals with the variant in that family presented with congenital cataract that were accompanied by a range of ophthalmological features but also leukoencephalopathy and stroke in some individuals,33 which may have important health implications for our family.
The start-loss variant in the LEMD2 gene presents an interesting finding in family CRVEEH79. LEMD2 was only recently confirmed as a cataract gene, following the identification of a p.(Leu13Arg) change in the LEM domain in families with autosomal recessive juvenile cataracts in the Hutterite community of North America.35 This previous report of the LEMD2 gene in relation to paediatric cataract by Boone et al35 found an additional relationship between variant carriers and sudden onset cardiac death, which may be pertinent to other individuals with LEMD2 variants. However, autosomal dominant disease has not previously been reported with this gene and functional investigation of the potential for cataract development with this start-loss variant is needed. Increased screening of this gene in cataract patients will also assist in informing inheritance trends and genotype-phenotype correlations.
The findings in family CSA93, with the c.386G>A p.(Arg129His) change in PRX must be interpreted with caution. Impacting the coding region of only the S-periaxin isoform, this variant would greatly value independent confirmation of pathogenic changes to lens function and cataract formation. While the PRX protein has been shown as important for lens fibre cell structure in mice36 the lack of cataract development and accompanying neurological features indicate this gene is still lacking the key evidence needed to confirm it causes congenital cataracts alone. This is further supported by PRX variants more often causing Charcot-Marie-Tooth disease (MIM:614895) and Dejerine-Sottas (MIM:145900), which are recessively inherited neurological conditions impacting the peripheral nervous system.
Variant interpretation continues to evolve with improved uptake in the reporting of cataract variants. Subsequently, our understanding of the disease-causing capacity of variants in cataract genes and their functional regions also continues to improve. This information critically underpins variant interpretation of newly identified variants using criteria such as the ACMG-AMP guidelines.15 Due to the rarity of the disease and the unique nature of the variants identified it would be expected that many families display variants classified as being of uncertain significance. This is compounded further by the breadth of genes associated with paediatric cataract that contribute to additional ocular and syndromic phenotypes. We have worked to stringently apply variant classification criteria and subclassify VUS based on their collective supporting evidence of pathogenicity. This has clearly identified those with the potential to reach likely pathogenic (or pathogenic) classification and are likely to account for the cataracts observed. Small family sizes frequently limited the use of cosegregation as a pathogenic evidence, but with variant reporting in databases such as ClinVar this may enable future reclassification with additional observations. The identification of these VUS in less-established cataract genes such as LEMD2 and PRX highlights the work that remains, in the research setting, to better understand the role they play in cataractogenesis. For VUS identified in well-established cataract genes, such as the connexins, a move towards gene-specific variant classification criteria for isolated paediatric cataracts would be advantageous, as would establishing functional assays for routinely assessing the functional effects of novel variants.
Of the subset of families being reassessed following the previous screen of 51 cataract genes, all variants identified in those probands were in genes not previously assessed, with the exception of the likely pathogenic HSF4 p.(Lys64Glu) variant in CSA168-01. All those variants received a VUS classification based on restrictions including inappropriate segregation or current limitations to our understanding of the gene, such as the capacity for autosomal dominant cataracts with variants in the LEMD2 gene. These do, however, provide informative observations that may inform our future understanding of these genes. The genome sequencing data will allow for periodic reassessment of newly identified genes and further evaluation of copy number and non-coding variants. Currently, there are conflicting reports of increased molecular diagnostic rates when using genome sequencing in congenital cataract cases.4 32 Our data currently indicate marginal difference with variants that would be equally identified with exome sequencing or a targeted gene panel. However, the full extent of the genomes sequencing benefits will be best measured in the coming years following the application of routine rescreening of genomic data in unsolved cases and an improved ability to identify and correctly interpret non-coding variants as pathogenic.
Data availability statement
Data are available on reasonable request. All variants reported in this study have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/); ClinVar accession numbers SCV001573165-SCV001573189. Additional data can be made available on request if within the ethical bounds of this study.
Ethics statements
Patient consent for publication
Ethics approval
The study was conducted according to the guidelines of the Declaration of Helsinki and followed the National Health and Medical Research Council statement of ethical conduct in research involving humans (2007, updated 2018). This study was approved by the Tasmania Health and Medical Human Research Ethics Committee (H0014539 14 Nov 2014 and 23875 12 Dec 2020), the Southern Adelaide Clinical Human Research Ethics Committee (3-07 19 Jul 2013 and 29 Nov 2019) and the Royal Victorian Eye and the Ear Hospital Human Research Ethics Committee (01/432H/11 6 Mar 2014). Study participants were recruited after they or their guardian gave written informed consent. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would like to thank the research participants involved in this study. We would like to thank Illumina and the Ramaciotti Centre for Genomics for the genome sequencing data obtained following receipt of the 2019 Genetic Society of Australasia student research grant schemes ‘Illumina NovaSeq Mini Grant’.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conceptualisation, KPB, JEC, DAM and JLJ; Methodology, JLJ, BJM, KPB and JCC; Software, BJM and JCC; Validation, JLJ; Formal analysis, JLJ and BJM; Investigation, JLJ, SES, ES and LSK; resources, JEC, DAM, JBR, JEE, DT, JP and TC; Data curation, BJM; Writing—original draft preparation, JLJ, KPB; Writing—review and editing, all; Visualisation, JLJ; Supervision, KPB and DAM; Project administration, ES, SES and LSK; Funding acquisition, KPB, JBR and DAM. Guarantor, KPB. All authors have read and agreed to the published version of the manuscript.
Funding This work was supported by a National Health and Medical Research Council (NHMRC) of Australia Ideas grant (GNT1185477) and Centre for Research Excellence grant (GNT1116360). JLJ was supported by the Australian Government Research Training Program and KPB was supported by a NHMRC Senior Research Fellowship. The Centre for Eye Research Australia receives Operational Infrastructure Support from the Victorian Government.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.