Article Text

Abstract
Objective We conducted a feasibility study to verify the effectiveness of 16S ribosomal RNA (rRNA) gene analysis using the nanopore sequencer MinION for identifying causative bacteria in several types of ocular infections.
Methods and Analysis Four cases of corneal ulcers, one case of endophthalmitis and one case of a conjunctival abscess were included in this study. DNA was extracted from corneal scraping, vitreous samples and secretions from the conjunctival abscess. We conducted 16S rRNA gene amplicon sequencing using MinION and metagenomic DNA analysis. The efficacy of bacterial identification was verified by comparing the conventional culture method with smear observations.
Results 16S rRNA gene sequencing analysis with MinION identified the causative organisms promptly with high accuracy in approximately 4 hours, from ophthalmic specimens. The results of the conventional culture method and 16S rRNA gene sequencing were consistent in all cases. In four of the six cases, a greater variety of organisms was found in the 16S rRNA gene analysis than in bacterial culture.
Conclusion Using our workflow, 16S rRNA gene analysis using MinION enabled rapid and accurate identification possible in various kinds of bacterial ocular infections.
- infection
- diagnostic tests/investigation
- microbiology
Data availability statement
Data are available in a public, open access repository. No data are available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known on this topic?
Analysis of partial 16S ribosomal RNA (rRNA) gene sequences by short-read sequencers requires a long period for bacterial identification. The nanopore sequencer, MinION, allows for a good resolution and rapid identification of bacteria.
What this study adds?
Our workflow using full length 16S rRNA gene analysis with MinION works well in identifying the causative organism of ocular bacterial infections from a small specimen amount.
How this study might affect research, practice or policy?
This new technique is a potential tool with high efficacy for the early diagnosis of ocular bacterial infections.
Introduction
Identification of pathogenic organisms is essential for the treatment of ocular infections. The gold-standard method is culturing on blood agar and/or smear examination. However, the culture of organisms requires several days, and often results in negative results when antibiotics are administered.1 2 Additionally, some bacteria are difficult to culture. In smear samples, exact identification of the organism is impossible, and some micro-organisms cannot be visualised by Gram staining. Furthermore, the colour and shape of organisms are often affected by antibiotics.
Genetic analyses have been developed and applied clinically to compensate for these shortcomings and to improve the accuracy and speed of identification.3–7 The 16S ribosomal RNA (rRNA) gene is a molecular marker that is predominantly used for bacterial classification.8 9 These genes are composed of conserved and variable regions, and gene sequencing using universal primers of the conserved region enables the identification of a wide range of bacteria. Gene analysis with the universal primers in 16S rRNA using a short-read sequencer has been applied in the analysis of ocular infectious diseases as well as intestinal microbiomes.10–17 However, the conventional short-read sequencer cannot yield reads covering the full length of the 16S rRNA gene, several regions of which have been targeted for sequencing,18 which often causes ambiguity in taxonomic classification.19 In addition, next generation sequencer (NGS) is usually based on several parallel fluorescence/proton scanning runs that obtain large amounts of nucleotide sequence data, but require days to weeks to complete.20
MinION, a long-read nanopore sequencer, overcomes the disadvantages of NGS and has been used in a variety of medical fields.21–24 We previously established a suitable workflow of 16S rRNA gene analysis using the MinION nanopore sequencer for bacterial identification.25 26 In this feasibility study, we applied this workflow of gene analysis using nanopore sequencing for various ocular infections.
Materials and methods
Subjects
Six eyes of six patients with ocular infections were studied, who visited the Department of Ophthalmology, Kansai Medical University Hospital, from 1 January 2020 to 30 September 2020. Patients were involved in the design, conducting, reporting and dissemination plans of our research.
Smear and culture method
Corneal specimens were obtained by scraping with a spatula and divided into three. Each specimen was applied for smear, culture and 16S rRNA gene analysis respectively. A 2.5 mL of vitreous fluid from patients with endophthalmitis was collected during vitrectomy from patients with endophthalmitis, and 0.1 mL of abscess contents from patients with conjunctival abscesses were used for conventional culture and 16S rRNA gene analysis. The details of each case are shown in figure 1. Smear samples were used for Gram staining. Each specimen was cultured on both blood and Sublow agar plates at 37°C under aerobic conditions. In case 2, Moraxella spp was suspected by culture, but the species could not be identified; therefore, matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF MS) was applied to identify the species.

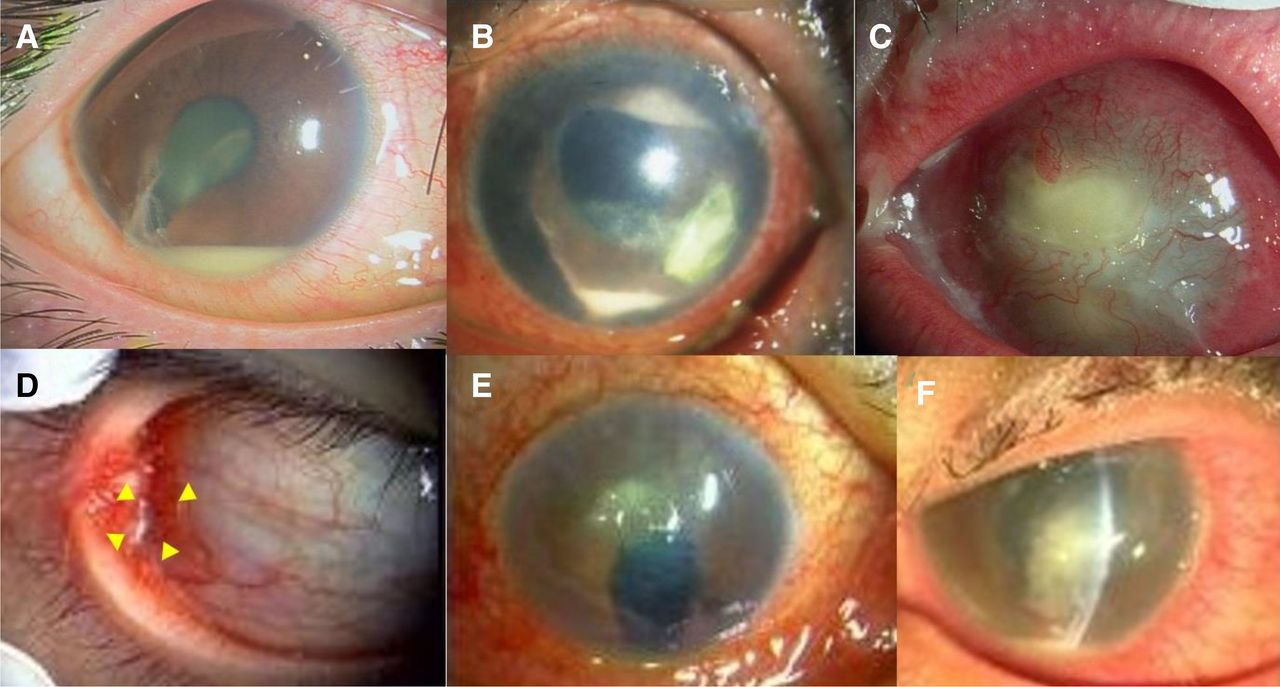
Photographs of entry cases. (a) Case 1: Traumatic endophthalmitis. (b) Case 2: Infectious keratitis. (c) Case 3: Infectious keratitis. (d) Case 4: Conjunctival abscess. (e) Case 5: Infectious keratitis. (f) Case 6: Infectious keratitis. Details are provided in Cases in Result section.
DNA extraction, PCR of 16S rRNA genes and sequencing
DNA extraction
Each ocular sample (corneal scraping, vitreous aspiration and secretion of conjunctival cyst) was used for conventional culture, smear and genetic analysis. The exact protocol for gene analysis has been previously described (dataset)26 Matsuo et al. Data from: Full-length 16S rRNA gene amplicon analysis of human gut microbiota using MinION nanopore sequencing confers species-level resolution. BMC Microbiol 2021. https//doi/10.1186 /s12866-021-02094-5. A detailed protocol is available at protocols.io (https://dx.doi.org/10.17504/protocols.io.bwr5pd86). Briefly, beads were added to the samples, which underwent bead-beating for 5 min using a vortex mixer. DNA was extracted using the Maxwell RSC Blood DNA Kit (Promega, AS1400) and used as a template to amplify bacterial 16S rRNA genes. PBS solution, which was used as a solvent for specimen collection, was processed according to the same protocol and used as a negative control.
PCR of 16S rRNA genes
The 16S rRNA gene was amplified using the KAPA2G Robust HotStart ReadyMix PCR Kit (KK5701; KAPA Biosystems, Wilmington, Massachusetts, USA) with the following primers: forward, 5'-TTTCTGTTGGTGCTGATATTGC AGRGTTYGATYMTGGCTCAG-3 and reverse, 5'-ACTTGCCTGTCGCTCTATCTTCCGGYTACCTTGTTACGACTT-3'. According to the manufacturer’s protocol, the barcodes were added to the amplified DNA by a second PCR using a PCR Barcoding Kit (SQK-PBK004; Oxford Nanopore Technologies, Oxford, UK). The amplification conditions were as follows: first PCR: initial denaturation at 94°C for 2 min; 35 cycles of 94°C for 15 s, 55°C for 15 s, and 68°C for 30 s; second PCR: initial denaturation at 95°C for 3 min; 35 cycles of 95°C for 15 s, 62°C for 15 s, and 72°C for 30 s. The amplicons were analysed by electrophoresis using 1.0% agarose gel.
Nanopore sequencing
Barcoded 16S rRNA gene amplicons were purified using AMPure XP (A63880, Beckman Coulter) and quantified using the QuantiFluor ONE dsDNA System (E4870, Promega). Equal amounts of amplicons were pooled (100 ng DNA in 10 µL), and the sequencing library was prepared according to the manufacturer’s instructions. For samples in which DNA concentration was lower than the detection limit of the device, we added the same volume of eluate as other samples pooled in the library construction. The library was sequenced using the Flongle flow cell R9.4.1 (FLO-FLG001, Oxford Nanopore Technologies) on a MinION Mk1B nanopore sequencer. Sequencing was continued until the number of output reads reached a plateau (approximately 4 hours).
Data analysis
Base-calling and adapter/barcode trimming were performed using Guppy software V.5.0.11 (Oxford Nanopore Technologies) to generate FASTQ-formatted sequence files. To eliminate reads outside the expected size range, the sequences were filtered by length using the SeqKit software V.0.10.0, retaining sequences between 1300 and 1950 bases. To identify bacteria, the processed reads were analysed using the cloud-based EPI2ME FASTQ 16S bacterial classification workflow V.2021.03.05, provided by Oxford Nanopore Technologies. The FASTQ files were uploaded using EPI2ME Desktop Agent V.3.3.0 (Oxford Nanopore Technologies), and the reads were aligned using BLAST against the NCBI 16S rRNA database containing 21 826 bacterial and archaeal strains. Each read was classified using the default settings, except for the minimum identity score, which was 85%. Low-abundance taxa with less than 1% classified reads were discarded from the analysis.
Results
The organisms identified by culture, smear and gene sequencing in all the cases are summarised in table 1. Except for case 4, no antibiotic eye-drops were prescribed before sampling.
Identified organisms by culture, smear and gene sequencing in each case
Cases
Case 1
An early 20s man was diagnosed with endophthalmitis of the right eye caused by an iron body attack. The radiograph depicted an intracameral foreign body with a high density. The patient underwent a vitrectomy with antibiotic perfusion (figure 1).
Case 2
An early 80s man presented with an infectious corneal ulcer in the left eye. The patient had a history of vitrectomy and silicone tamponade for proliferative vitreoretinopathy.
Case 3
A late 80s woman with infectious keratitis presented with bullous keratopathy in the left eye.
Case 4
An early 40s man presented with an ocular conjunctival abscess in the right eye. He was treated with antibiotics; however, the abscess did not improve.
Case 5
A late 80s man was a hard contact lens wearer with an aphakic eye in his right eye due to childhood trauma. He had hyperaemia and ocular discharge, and was diagnosed with a corneal ulcer.
Case 6
An early 40s man was found to be an inappropriate soft contact lens wearer. He had a corneal ulcer with pain, hyperaemia and ocular discharge in the left eye.
In each of the six cases, there was good concordance between the most abundant organism identified by 16S rRNA gene sequencing and culture.
Some differences were found between gene analysis and conventional culture methods. In case 2, Moraxella was identified in the bacterial culture at the genus level. Species-level classification (Moraxella nonliquefaciens) was achieved by MinION sequencing, and the results were consistent with those obtained by MALDI-TOF MS. In cases 1, 4, 5 and 6, more organisms were found in the gene analysis than in the culture. For example, in case 5, Pseudomonas aeruginosa was most dominant in the genetic analysis of MinION, and was also detected in the culture. Additionally, gene analysis using MinION detected Peptoniphilus lacydonensis, Cutibacterium acnes and Staphylococcus epidermidis at low frequencies. In case 6, Candida spp was also detected by blood agar culture and P. aeruginosa, which was detected by gene analysis and culture. Meanwhile, smear examination could detect the organism in only four cases (cases 2, 4, 5 and 6).
The negative control generated a small number of reads assigned to Ralstonia pickettii, which was considered a typical example of potential background contamination.27
Discussion
Culture-based techniques have been the standard for detecting pathological microorganisms in clinical specimens. Metagenomic sequencing has emerged as an alternative methodology for overcoming the drawbacks of conventional bacterial cultures. 16S rRNA gene amplicon sequencing is useful for identifying causative bacteria from clinical samples with a relatively low bacterial load, such as ophthalmological specimens.
Metagenomic analysis with NGS platforms has been reported to identify Achromobacter in a case of scleral buckle infection, Corynebacterium propinquum, in contact lens-related microbial keratitis,14 15 and can also be applied for detecting unculturable bacterial pathogens. In a clinical context, this technique is useful, especially for identifying causative agents that are difficult to grow in culture, such as nutritionally variant streptococci, even after antibiotic administration.28 29 However, such short-read NGS technology has some disadvantages in terms of speed and accuracy, which leads to low taxonomic resolution and time consumption for bacterial identification. The portable nanopore sequencer MinION used in this study can analyse long DNA sequences covering the full-length bacterial 16S rRNA gene. Our previous study showed that full-length 16S rRNA gene amplicon sequencing with MinION provides a higher taxonomic resolution than short-read sequencing using the short-read NGS platform.26 Accumulating evidence demonstrates promising results with MinION sequencing, which enables bacterial identification at the species level.8 Another intriguing feature of MinION sequencing is that it generates sequencing reads in real-time, allowing for a far shorter turnaround time for data processing. In our approach, bacterial identification of clinical specimens could be completed within a total analysis time of approximately 4 hours in the shortest time (figure 2). This would offer a significant advantage for the rapid diagnosis of infectious diseases.

{kind=link}
{kind=link}
Schema of the platform of 16S rRNA gene amplification analysis with MinION. It takes at least 4 hours from sample collection to the sequencing.
Given the excellent discriminatory power of MinION long-read sequencing, we validated its efficacy in identifying pathogenic bacteria in a wide range of ophthalmological samples. MinION sequencing successfully identified bacterial species from the vitreous fluid, corneal scrapes and abscess samples. The profiles of dominant taxa analysed by MinION sequencing were consistent with those obtained from the bacterial cultures in all samples tested. As for Moraxella, species-level identification is not attainable by culture-based methods, and mass spectrometry has been utilised for accurate taxonomic classification. We successfully identified M. nonliquefaciens by MinION sequencing, which was confirmed by MALDI-TOF MS analysis. In case 6, both fungi and P. aeruginosa were detected in the culture. Fungi were suspected to be the primary causative organisms, which may explain the low abundance of bacteria estimated from the 16S rRNA gene sequencing results. Although our experimental platform is limited to identifying bacteria, nanopore amplicon sequencing also allows the identification of pathogenic fungi by using universal primer sets that target a genetic marker, such as the internal transcribed spacer of the fungal rRNA gene.
These results suggest that full-length 16S rRNA gene amplicon sequencing via MinION is a reliable and practical tool for the rapid identification of pathogenic bacteria with high resolution in ocular specimens. All the identified organisms matched the culture, smear, or clinical findings in each case. Thus, we have made the decision of causative organisms from these various kinds of analyses.
Low et al reported 16S rRNA gene analysis using a portable nanopore sequencer in two cases of keratitis.30 Jun et al also used a nanopore sequencer in eight cases of endophthalmitis.31 Low et al reported the advantages of gene analysis using MinIONin detecting causative organisms after administering antimicrobial eye drops. Jun et al described the advantage of gene analysis using MinION for the early detection of endophthalmitis in the anterior aqueous humour at the first visit. In both reports, the number of cases was small, and the authors emphasised the need for a greater number of cases.
Low et al stated that the collection of keratitis specimens is affected by the material and shape of the swabs used. In this study, we used a direct spatula to scrape the ulcerative tissue, providing sufficient DNA for analysis. The amounts of the specimens from conjunctival abscesses and endophthalmitis were small, that is, 0.1 mL of leaking purulent material and 2.5 mL of aspirated vitreous. However, we were able to detect the causative organism in both cases. The primer sequence for the bacteria used by Jun et al is unclear, but the kit manual they used indicates that some types of bacteria may not be detectable. In contrast, we used a primer set optimised to amplify the 16S rRNA genes across a broad range of bacterial species, the specificity of which has been confirmed in the analysis of the intestinal microflora.26 We used the database provided by Oxford (EPI2ME Fastq16S), as in Jun et al.
Genetic analysis using MinION has some limitations. First, compared with conventional methods such as MiSeq, MinION might be less accurate in determining individual bases and have a higher error rate. However, this can be compensated by long-chain decoding. Second, as was seen in cases 5 and 6, it is often the case that a variety of bacteria are detected. Therefore, the possibility of contamination by indigenous microorganisms should be carefully considered. In a review of diagnostic methods for ocular infections, Eguchi also stated that the diversity of micro-organisms detected by genetic analysis requires careful analysis to determine whether they are contaminated or indigenous microorganisms.32 Due to the high sensitivity of amplicon sequencing, even a small amount of DNA contaminated during the sample preparation can lead to false-positive results. For reliable detection of pathogenic bacteria, it is required to minimise the risk of DNA contamination both in collecting clinical specimens and the subsequent experimental procedures. Furthermore, common background species should be carefully distinguished from infectious pathogens. Especially in a low-level infection or a sample with low read counts, the presence of the contaminants makes it difficult to interpret the sequencing results. Although it is challenging to completely rule out the influence of these contaminants, incorporating negative controls processed precisely the same way should be a minimum requirement to ensure the validity of the results and distinguish contaminants from true pathogens. Third, taxonomic resolution is only at the species level and not at the strain level. Therefore, virulence factors cannot be identified, and genetic analysis cannot differentiate dead micro-organisms from live ones.
Even if these factors are considered, MinION could provide high resolution and rapid identification from a small specimen amount, and we believe that it will contribute greatly to daily clinical practice.
Conclusion
Using our workflow, 16S rRNA gene analysis using MinION enabled rapid and accurate identification possible in various kinds of bacterial ocular infections.
Data availability statement
Data are available in a public, open access repository. No data are available.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the Ethical Committee of Kansai Medical University (approval no. 2020186). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We are grateful to the PhD, Keiko Toyama, for her technical support. We would like to thank Editage (www.editage.com) for English language editing.
References
Footnotes
Contributors MO and YM contributed equally to this paper. All authors made substantial contributions to the design and analysis of this work. MO, KS, YM, and KH, contributed to the conception and design of the study, data analysis, and drafting of the manuscript. MO, KS, SO, and HY contributed to the data acquisition. KT and KH contributed as the project supervisors. All the authors approved the final manuscript.
Funding AMO (Johnson & Johnson, Japan) grant support.
Competing interests None declared.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.