Article Text

Abstract
Aim To determine the proportion of idiopathic orbital inflammation (IOI) and orbital benign lymphoid hyperplasia (OBLH) accounted for by immunoglobulin (Ig)G4-related orbital disease (IgG4-ROD) using the comprehensive diagnostic criteria for IgG4-related disease published by Umehara et al and the consensus diagnostic criteria published by Deshpande et al. Secondary aims were to compare the histological and clinical features of IgG4-ROD and non-IgG4-ROD cases, and to compare IgG4-ROD cases diagnosed using the comprehensive diagnostic criteria with those diagnosed using the consensus diagnostic criteria.
Methods A retrospective histopathological review and clinical case series. 55 cases of biopsy-confirmed non-granulomatous IOI and 10 cases of biopsy-confirmed OBLH were included. The intensity of sclerosis, lymphoplasmacytic infiltration and eosinophilic infiltration was graded from 0 to 3+ using a standardised and validated scoring system.
Results IgG4-ROD accounted for 50% and 40% of cases originally diagnosed as OBLH and 23.6% and 5.4% of cases originally diagnosed as IOI, using the comprehensive diagnostic criteria and the consensus diagnostic criteria, respectively. IgG4-ROD cases had numerous significant histological differences, but relatively few significant clinical differences, from non-IgG4-ROD cases. Compared with the comprehensive diagnostic criteria, the consensus diagnostic criteria identified a group of IgG4-ROD cases with a slightly higher ratio of IgG4+ to IgG+ (p=0.01) and a slightly longer duration of symptoms (p=0.02).
Conclusions This is the largest review of IgG4 staining among biopsy-confirmed IOI and OBLH. IgG4-ROD accounted for a substantial proportion of OBLH. The prevalence among cases of IOI was considerably reduced when the consensus diagnostic criteria were used in place of the comprehensive diagnostic criteria.
- Orbit
- Pathology
- Lacrimal gland
- Inflammation
Statistics from Altmetric.com
Introduction
Idiopathic orbital inflammation (IOI) and orbital benign lymphoid hyperplasia (OBLH) are benign, idiopathic, tumefactive conditions of the orbit. Together, they have been reported to account for 30%–70% of all orbital biopsies.1 ,2 IOI is a non-infectious inflammation of the orbital soft tissues that cannot be attributed to another disorder. It is a diagnosis of exclusion and comprises cases with heterogeneous clinicopathology. As diagnostic techniques improve, lesions are being classified out of this idiopathic category and the term's scope is contracting. Benign lymphoid hyperplasia (LH) is a benign, mass forming proliferation of polyclonal lymphocytes without monoclonality or other evidence of neoplasia. These cases may have an inflammatory presentation and, like IOI, are thought to represent an abnormal immune response to some unknown stimulus.3
Immunoglobulin (Ig)G4-related disease (IgG4-RD) is a systemic disorder characterised by soft tissue mass lesions infiltrated with IgG4-bearing plasma cells. It unites a wide spectrum of conditions that were previously labelled as idiopathic. In the orbit, lesions begin as hypercellular lymphoplasmacytic masses with morphology not dissimilar to OBLH. Over time, lesions may become progressively sclerosed and more resemblant of IOI.4 ,5 Cases are likely to be misdiagnosed as OBLH or IOI if IgG4 staining is not performed to reveal the diagnosis. Assigning a diagnosis of IgG4-RD has significance for the patient as it implies an increased risk of IgG4-RD affecting other organs, and possibly an increased risk of malignancy, particularly non-Hodgkin lymphoma.6 ,7 The prevalence of IgG4-related orbital disease (IgG4-ROD) is difficult to estimate as published studies: (1) have been few in number; (2) used different criteria for diagnosing IgG4-RD; (3) tended to enrol small groups of patients with heterogeneous pathology; and (4) have been of uncertain generalisability as most reports have come from Asian centres and the presence of racial or geographic variation in the frequency of IgG4-ROD is unknown.6 ,8–14
The primary aim of this study was to retrospectively determine the proportion of biopsy-confirmed IOI and OBLH accounted for by IgG4-ROD in an Australian population using both the comprehensive diagnostic criteria for IgG4-RD published by Umehara et al,15 and the consensus diagnostic criteria for IgG4-RD published by Deshpande et al16 (box 1). Secondary aims were to: (1) compare the histological features of the IgG4-ROD and non-IgG4-ROD cases using a standardised histology grading system, (2) compare the clinical features of the IgG4-ROD and non-IgG4-ROD cases and (3) evaluate whether using different consensus diagnostic criteria alters the collective phenotype of the cases diagnosed as IgG4-ROD.
Diagnostic criteria for IgG4-related orbital disease (ROD)
Comprehensive diagnostic criteria for IgG4-related disease by Umehara et al15
-
Clinical examination showing characteristic diffuse/localised swelling or masses in single or multiple organs.
-
Haematological examination shows elevated serum IgG4 concentration (≥135 mg/dL).
-
Histopathological examination shows:
-
Marked lymphocyte and plasma cell infiltration and fibrosis
-
Infiltration of IgG4+ plasma cells: ratio of IgG4+ to IgG+ cells >40% and >10 IgG4+ plasma cells/high power field (HPF)
Definite: (1)+(2)+(3); probable: (1)+(3); possible: (1)+(2)
-
Consensus diagnostic criteria for IgG4-ROD by Deshpande et al16
-
Lacrimal gland biopsy demonstrates 1 of the following or orbital biopsy demonstrates ≥2 of the following:
-
Dense lymphoplasmacyic infiltrate
-
Fibrosis, usually storiform in character
-
Obliterative phlebitis
-
-
Ratio of IgG4+ to IgG+ cells > 40% and >100 IgG4+ plasma cells/HPF
Highly suggestive of IgG4-related disease: (1)+(2).
Patients and methods
This study was a retrospective histopathology review and clinical case series of patients with biopsy-confirmed IOI and OBLH. Institutional Ethics Committee approval was obtained.
Patients
Cases were identified by searching the specimen archive of South Australian pathology providers from the period 1 January 1999 to 31 December 2012. All cases with a histopathological diagnosis of IOI or OBLH were retrieved. The histopathology slides and clinical records were reviewed to confirm or refute the original diagnosis. The diagnosis of OBLH was given to specimens that exhibited a benign, follicular lymphoid infiltrate with scant or absent fibrosis. The presence of ‘tingible body’ macrophages was supportive of the diagnosis.17 Exclusion criteria included: (1) conjunctival biopsies (a site reported to be involved by IgG4-RD very rarely);6 ,18 (2) clinical or biochemical features suggestive of thyroid eye disease; (3) granulomatous inflammation; (4) evidence of monoclonality on immunophenotyping or PCR; and (5) insufficient tissue in the specimen block to perform IgG and IgG4 immunostaining.
Immunohistochemistry for IgG and IgG4
For each case, two adjacent 4-μm thick, formalin-fixed, paraffin embedded sections with the highest intensity lymphoplasmacytic infiltrate were selected for immunohistochemical staining. The Benchmark XT automated slide stainer (Ventana Medical Systems, Tucson, Arizona, USA) was used to stain one slide for IgG and the other for IgG4. Both slides were treated with the ultraView detection kit (Ventana Medical Systems) followed by Haematoxylin 2 (Ventana Medical Systems) and Bluing reagent (Ventana Medical Systems).
Specimen assessment
Two pathologists independently reviewed all specimens and collaborated to reach a consensus whenever there was a discordant result. Both pathologists used high power field (HPF) areas of 0.196 mm2. For each specimen, three non-overlapping HPFs with the highest concentration of IgG4+ cells were selected and the number of IgG4+ cells and IgG+ cells were counted. The average number of IgG4+ cells per HPF and the average ratio of IgG4+ to IgG+ cells were recorded.
The intensities of the following histopathology features were graded from 0 to 3+: sclerosis, lymphoplasmacytic infiltration and eosinophilic infiltration. The grading system was standardised by defining the characteristics indicative of each score and by using a series of reference photomicrographs against which cases were judged. Sclerosis was categorised as ‘collagenous’ when it had a lamellar architecture and as ‘storiform’ when it formed a whirling pattern. The presence or absence of the following features was recorded: germinal centres, obliterative phlebitis and lymphoepithelial lesions. We defined lymphoepithelial lesions as aggregates of 3 or more lymphocytes within glandular epithelium, which is an adaptation of a previously published definition.19 The grading system was validated within our institution. A detailed description of the grading system and the validation protocol is outside the scope of this article and will be submitted for publication separately.
Diagnostic criteria for IgG4-ROD
Cases were categorised into IgG4-ROD and non-IgG4-ROD using both the comprehensive diagnostic criteria for ‘definite’ or ‘probable’ IgG4-RD published by Umehara et al15 and the consensus diagnostic criteria for IgG4-RD published by Deshpande et al (box 1).16
Clinical information
A chart review was undertaken for all cases. The following information was retrieved: demographic information (age, gender); medical history (atopic or autoimmune disease); presenting clinical features; laboratory data (serum IgG4, blood eosinophilia); radiological data (orbital and extra-orbital soft tissue swelling(s) radiologically); management data (treatment, response); and follow-up data (development of lymphoma, duration of follow-up). To maximise the validity of the results, clinical features were recorded as ‘present’ only if they were clearly documented as so, as ‘absent’ only if they were clearly documented as so, and as ‘data not recorded’ if neither of the previous criterion was fulfilled. The absence of documentation was not considered evidence of absence of the feature.
Statistical analysis
The clinical and histopathology features of the IgG4-ROD and non-IgG4-ROD groups were compared using the unpaired Student's t test (2-tailed) for continuous variables, Fisher's exact test (2-tailed) for categorical variables and the Mann–Whitney test for interval variables (histopathology scores). Statistical significance was defined as p<0.05. In the analysis of histological features, 95% CIs were calculated for the risk ratio (categorical variables) and for the difference between the means (continuous and interval variables).
Results
A total of 55 cases of IOI from 54 patients and 10 cases of OBLH from 9 patients fulfilled the inclusion and exclusion criteria for this study. The proportion of cases assigned a diagnosis of IgG4-ROD is listed in table 1. Using the comprehensive diagnostic criteria,15 18 orbital biopsies from 17 patients were ‘definite IgG4-RD’ (2 cases) or ‘probable IgG4-RD’ (16 cases). In all, 14 (77.8%) of these biopsies were from the lacrimal gland. All of the ‘probable IgG4-RD’ cases were relegated to that category because serum IgG4 had not been tested. Using the consensus diagnostic criteria,16 7 orbital biopsies from 7 patients were ‘highly suggestive’ of IgG4-RD; 5 (71%) of these biopsies were from the lacrimal gland. One of these 7 IgG4-RD patients had undergone repeat biopsy of the same lacrimal gland 7 years later, and the second biopsy demonstrated only 22.3 IgG4+ cells/HPF.5 This second biopsy, since it did not fulfil the consensus diagnostic criteria for IgG4-RD (albeit a false negative result), was counted as a non-IgG4-RD case for the purpose of the histological analysis. For the clinical analysis, however, the clinical features of the patient at the time of both biopsies were recorded in the IgG4-RD group. For this reason, the consensus diagnostic criteria resulted in 7 cases of IgG4-ROD for the histological analysis but 8 cases of IgG4-ROD (from 7 patients) for the clinical analysis.
Proportion of cases assigned a diagnosis of IgG4-related orbital disease (ROD)
Histopathology and laboratory features
The result of the IgG4 and IgG immunohistochemical stains is illustrated as a scatter plot in figure 1. Orbital inflammations were not organised into discrete IgG4+ and IgG4- groups but spanned a continuum from no IgG4 staining to intense IgG4 staining. A comparison of the histopathology features of the IgG4-ROD cases and non-IgG4-ROD cases is provided in table 2. Both sets of diagnostic criteria found that IgG4-ROD had significantly higher lymphoplasmacytic infiltration, germinal centre formation and blood eosinophilia at the time of orbital biopsy. The comprehensive diagnostic criteria, but not the consensus diagnostic criteria, found that IgG4-ROD had significantly more sclerosis (p=0.01) and eosinophilic infiltration (p=0.002). The character of sclerosis was storiform in just one case of IgG4-ROD. No case demonstrated obliterative phlebitis. The intensity of IgG4 staining was the only statistically significant histological difference between IgG4-ROD cases identified using each set of diagnostic criteria.
Comparison of IgG4-related orbital disease (ROD) and non-IgG4-ROD cases: histopathology

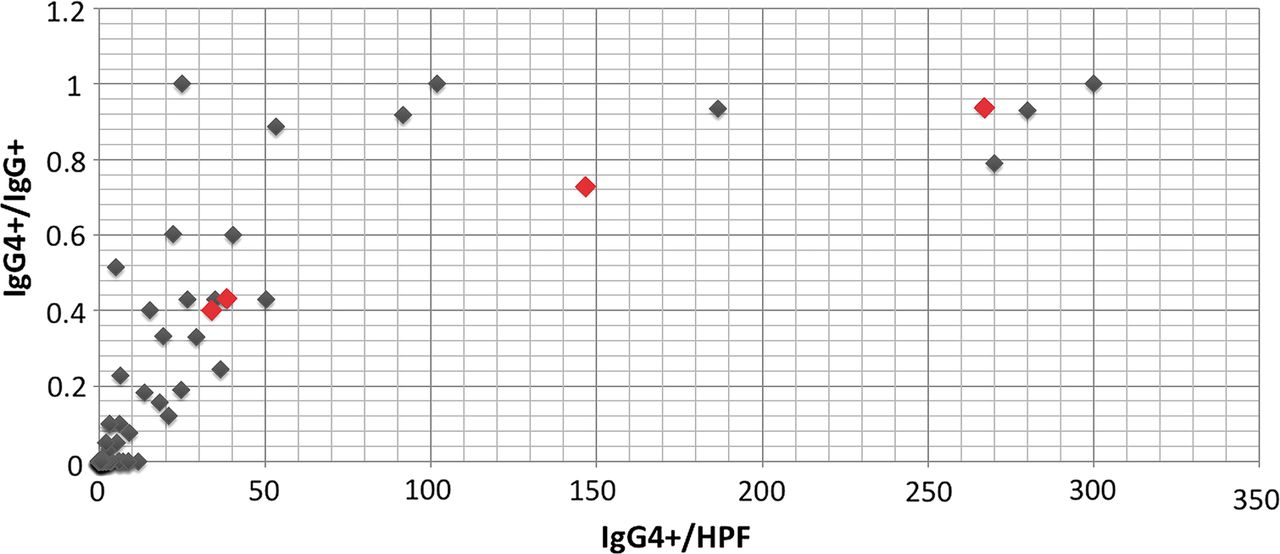{kind=link}
Scatter plot illustrating the number of IgG4+ cells/high power field (HPF) and the ratio of IgG4+ to IgG+ for 65 non-specific orbital inflammations. Each case appears as a single diamond. Red diamonds indicate cases of IgG4-related orbital disease diagnosed from biopsy of non-lacrimal gland orbital tissue. The cluster of diamonds in the bottom left corner comprises 39 cases.
Demographic, clinical and radiological features
The demographic, clinical and radiological features of IgG4-ROD and non-IgG4-ROD cases are presented in table 3. Although IgG4-ROD was clinically heterogeneous, the majority of cases presented with an insidious or subacute onset of symptoms related to orbital mass effect. Inflammatory signs tended to be mild. Presenting features of IgG4-ROD included eyelid or periocular swelling (78%–100%), proptosis (56%–75%), tenderness (25%–36%), dystopia (25%–33%), conjunctival or eyelid erythema (13%–25%), diplopia (13%–33%), and pain (0%–33%). Periorbital structures involved by IgG4-ROD on imaging included the lacrimal gland(s) (75%–77%), orbital fat (44%–50%), extraocular muscle(s) (33%–63%), orbital nerves (11%–13%), temporal fossa (11%–13%) and sclera (6%–13%).
Comparison of IgG4-related orbital disease (ROD) and non-IgG4-ROD patients: demographic, clinical and radiological data
Compared with non-IgG4-ROD, both sets of diagnostic criteria found IgG4-ROD cases to have a significantly higher rate of abnormal extra-orbital soft tissue swelling on imaging, most commonly involving the major salivary glands and cervical and mediastinal lymph nodes. The only statistically significant clinical difference between cases of IgG4-ROD selected using each set of diagnostic criteria was the duration of symptoms at the time of first review by an ophthalmologist (p=0.02). IgG4-ROD cases were highly heterogeneous in disease severity and course, and the response to corticosteroid treatment did not significantly differ from the non-IgG4-ROD group.
Discussion
This study is the largest review of IgG4 staining among cases of biopsy-confirmed IOI and OBLH. IgG4-ROD accounted for a large proportion of cases originally diagnosed as OBLH (50% with the comprehensive diagnostic criteria vs. 40% with the consensus diagnostic criteria) and a variable proportion of cases originally diagnosed as IOI (23.6% with the comprehensive diagnostic criteria vs. 5.4% with the consensus diagnostic criteria). IgG4-ROD had numerous significant histological differences, but relatively few significant clinical differences, from non-IgG4-ROD. Compared with the comprehensive diagnostic criteria, the consensus diagnostic criteria dramatically reduced the proportion of IOI cases diagnosed as IgG4-ROD. The consensus diagnostic criteria also identified IgG4-ROD cases with a longer duration of symptoms (p=0.02) and a higher intensity of IgG4 staining. There were no other significant histological or clinical differences between IgG4-ROD cases identified using each set of diagnostic criteria.
Previously published studies have employed a variety of different IgG4-RD diagnostic criteria, but comparisons can be drawn by interrogating the data retrospectively. Excluding conjunctival biopsies and applying the diagnostic thresholds of the comprehensive diagnostic criteria (IgG4/HPF >10, IgG4:IgG >0.4), IgG4-ROD has been found to account for: 4 of 62 (6.5%) cases of IOI, LH and lymphoma;12 18 of 78 (23%) cases of LH and lymphoma;9 5 of 8 (62.5%) ‘benign lymphoid lesions’;20 and 4 of 60 (6.7%) ‘orbital lesions biopsied due to clinical suspicion of lymphoma’.11 Two other studies investigated the proportion of IgG4-ROD among IOI and LH but unfortunately reported incomplete results to satisfy any current consensus-based IgG4-RD diagnostic criteria.13 ,21 Only one previous study has investigated the proportion of orbital IgG4-ROD among a single pathological entity (IOI), finding IgG4-ROD in 9 of 25 (36%) cases.14
Storiform fibrosis and obliterative phlebitis were rare in our series, in keeping with the findings of a 2012 meta-analysis of all published IgG4-ROD cases.6 Although storiform fibrosis and obliterative phlebitis are ubiquitous in most extra-orbital manifestations of IgG4-ROD, they should be considered organ-specific rather than disease-specific features. Interestingly, Deschamps et al14 reported a significant rate of non-obliterative phlebitis in their series of IgG4-ROD. This finding supports our theory that the low rate of obliterative phlebitis in IgG4-ROD may be an artefact of the tiny orbital veins being difficult to identify once obliterated.8
One important difference between the comprehensive and the consensus diagnostic criteria for orbital IgG4-RD is the intensity of IgG4 staining required for diagnosis (>10 vs.>100 IgG4+ plasma cells/HPF). Figure 1 demonstrates that orbital inflammations are not organised into discrete IgG4+ and IgG4− groups, but span a continuum from no IgG4 staining to intense IgG4 staining. The intensity of IgG4 staining that should be considered diagnostic of IgG4-ROD is therefore not self-defining. IgG4-RD is thought to be a distinct entity with a unique aetiology; however, IgG4 elevation is not specific for IgG4-RD,22 and there is a remote possibility that IgG4-RD comprises a heterogeneous group of disorders that happen to share a common immunological reaction pattern. Nevertheless, if one assumes that IgG4-ROD is a distinct entity, then the ideal diagnostic level would distinguish a group of cases with unique clinical and histological features. This study identified few clinical features that distinguished IgG4-ROD from non-IgG4-ROD, suggesting that morphological features must be relied upon to guide the diagnosis of IgG4-ROD. Using a diagnostic level of 10 IgG4+ cells/HPF, the IgG4-ROD cases had significantly higher intensities of sclerosis, lymphoplasmacytic infiltrate, eosinhophilic infiltrate and germinal centre formation. Statistical significance across all four parameters is preserved with a diagnostic level of 30 IgG4+ cells/HPF or 50 IgG4+ cells/HPF (data not shown), but not when a diagnostic level of 100 IgG4+ cells/HPF is used. As we have previously noted, obliterative phlebitis and storiform fibrosis are rare in IgG4-ROD and therefore cannot be relied upon as diagnostic adjuncts.8 Further studies are needed to validate diagnostic criteria for IgG4-RD affecting the orbit.
Although this study revealed useful insights into IgG4-ROD, we acknowledge several limitations. First, we looked for statistically significant differences between cases of IgG4-ROD and a mixed group comprising cases of IOI and cases of OBLH. It could be argued that IOI and LH are different conditions that should not be combined to form a single comparison group. Conversely, we feel that the terms ‘IOI’ and ‘OBLH’ simply describe tissue reaction patterns, rather than defining unique conditions, and therefore combining these cases does not compromise the validity of the results. Second, extra-orbital imaging and serum IgG4 testing was performed in relatively few cases, as most patients were managed prior to widespread awareness of IgG4-RD. Consequently, only 2 cases could be classified as ‘definite’ IgG4-RD according to the comprehensive diagnostic criteria. Third, complete clinical data were unavailable for a minority of cases. And, last, our exclusion criteria did not list features of granulomatosis with polyangiitis (GPA) other than granulomatous inflammation (such as necrosis or neutrophil infiltration). GPA may mimic IgG4-RD and the diagnosis should be considered in any IgG4-positive specimen demonstrating necrosis, microabscess formation or vasculitis.23
Conclusions
IgG4-ROD accounted for a large proportion of OBLH and a variable proportion of IOI, depending on whether the comprehensive or the consensus diagnostic criteria for IgG4-RD were applied. Cases of IgG4-ROD diagnosed using the consensus diagnostic criteria had a higher intensity of IgG4 staining and a longer duration of symptoms than cases identified using the comprehensive diagnostic criteria. IgG4 staining should be performed on all benign, non-granulomatous orbital biopsies as the diagnosis may have implications for the patient's systemic health.
References
Footnotes
-
Contributors Design of the study: NHA, NS, DJK and DS; data collection: NHA, NS and DJK; data analysis: NHA and DS; data interpretation: NHA and DS; manuscript preparation: NHA, NS, DJK and DS; manuscript review: NHA, NS, DJK and DS.
-
Competing interests None.
-
Ethics approval The Royal Adelaide Hospital Human Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement The unpublished raw data from this study are available from the authors of this manuscript.
Linked Articles
- At a glance