Mutation-Dependent Pathomechanisms Determine the Phenotype in the Bestrophinopathies

 , and
, and
Abstract
:1. Introduction
2. Results
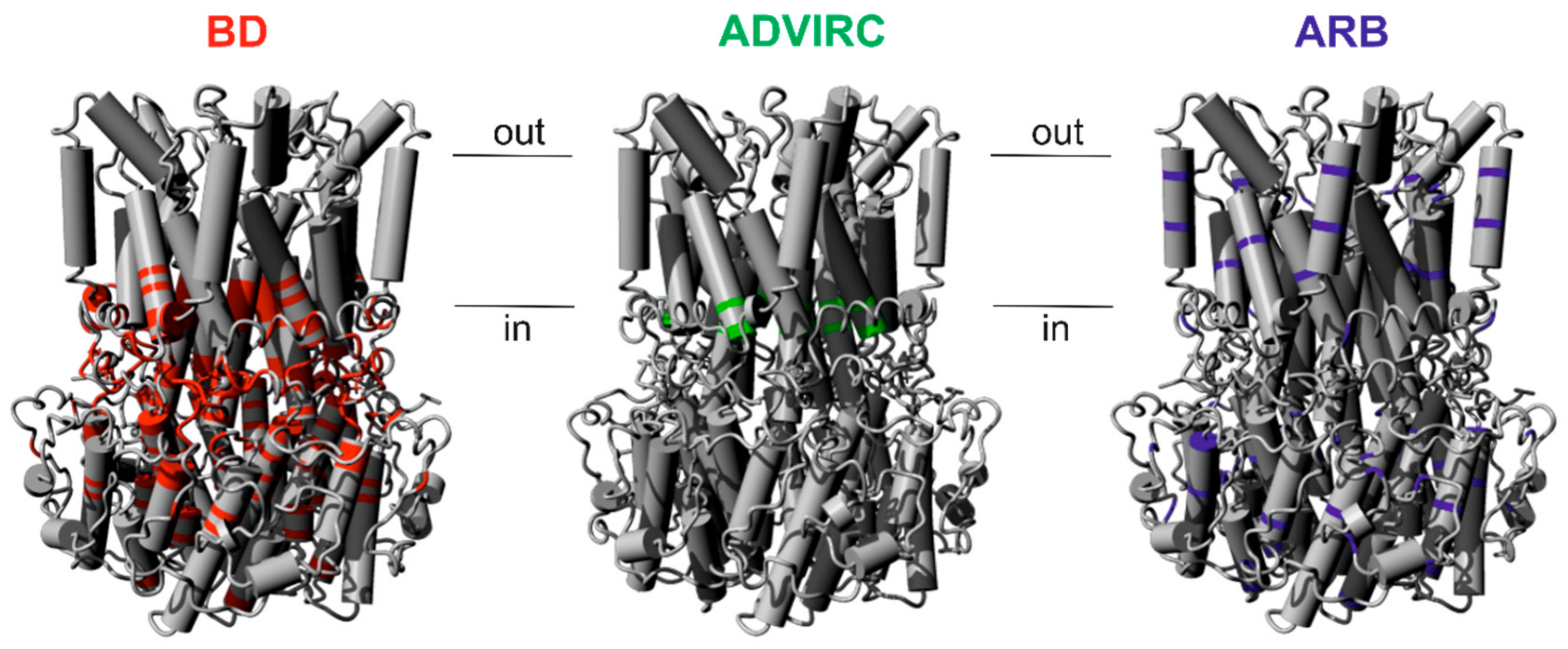
2.1. Assigning Best Vitelliform Macular Dystrophy (BD)-, Autosomal Dominant Vitreoretinochoroidopathy (ADVIRC)-, and Autosomal Recessive Bestrophinopathy (ARB)-Associated Mutations to the BEST1 Chicken Three-Dimensional (3D) Crystal Structure
2.2. In-Vitro Model of BD, ADVIRC, and ARB
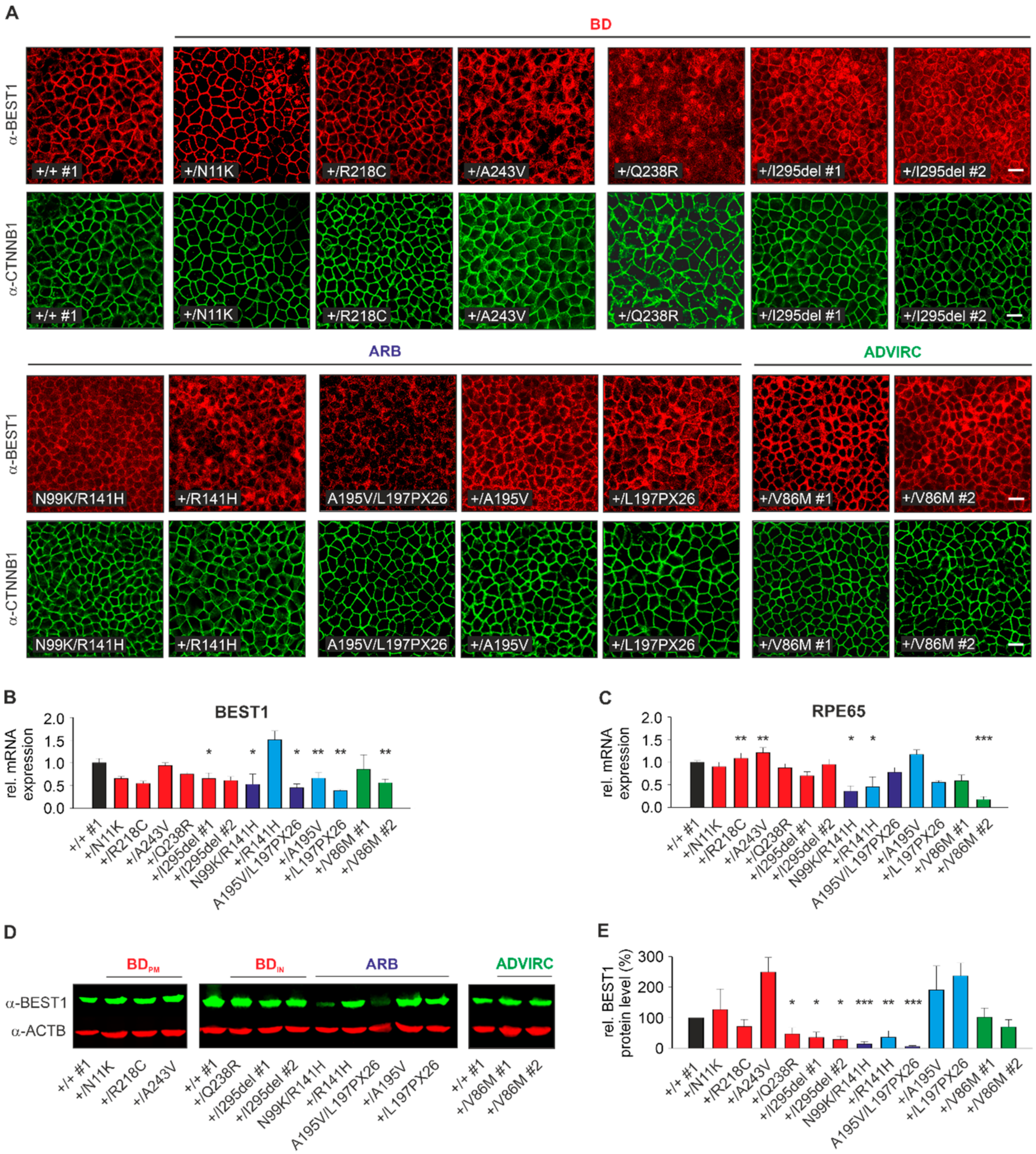
2.3. Localization, RNA, and Protein Expression of Mutant BEST1 in BD, ADVIRC, and ARB hiPSC-RPEs
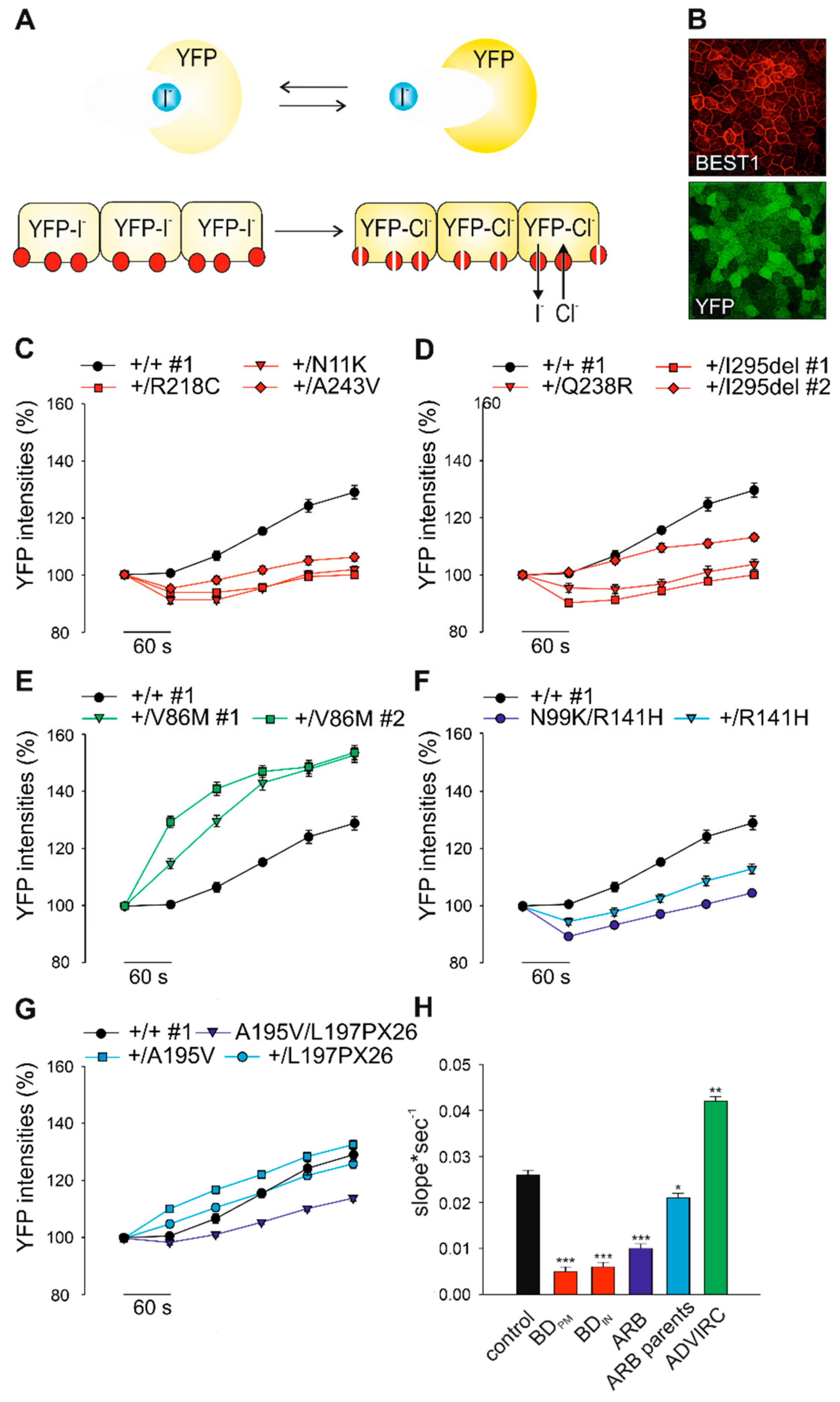
2.4. Effect of BD-, ADVIRC-, and ARB-Associated Mutations on BEST1-Mediated Anion Permeability
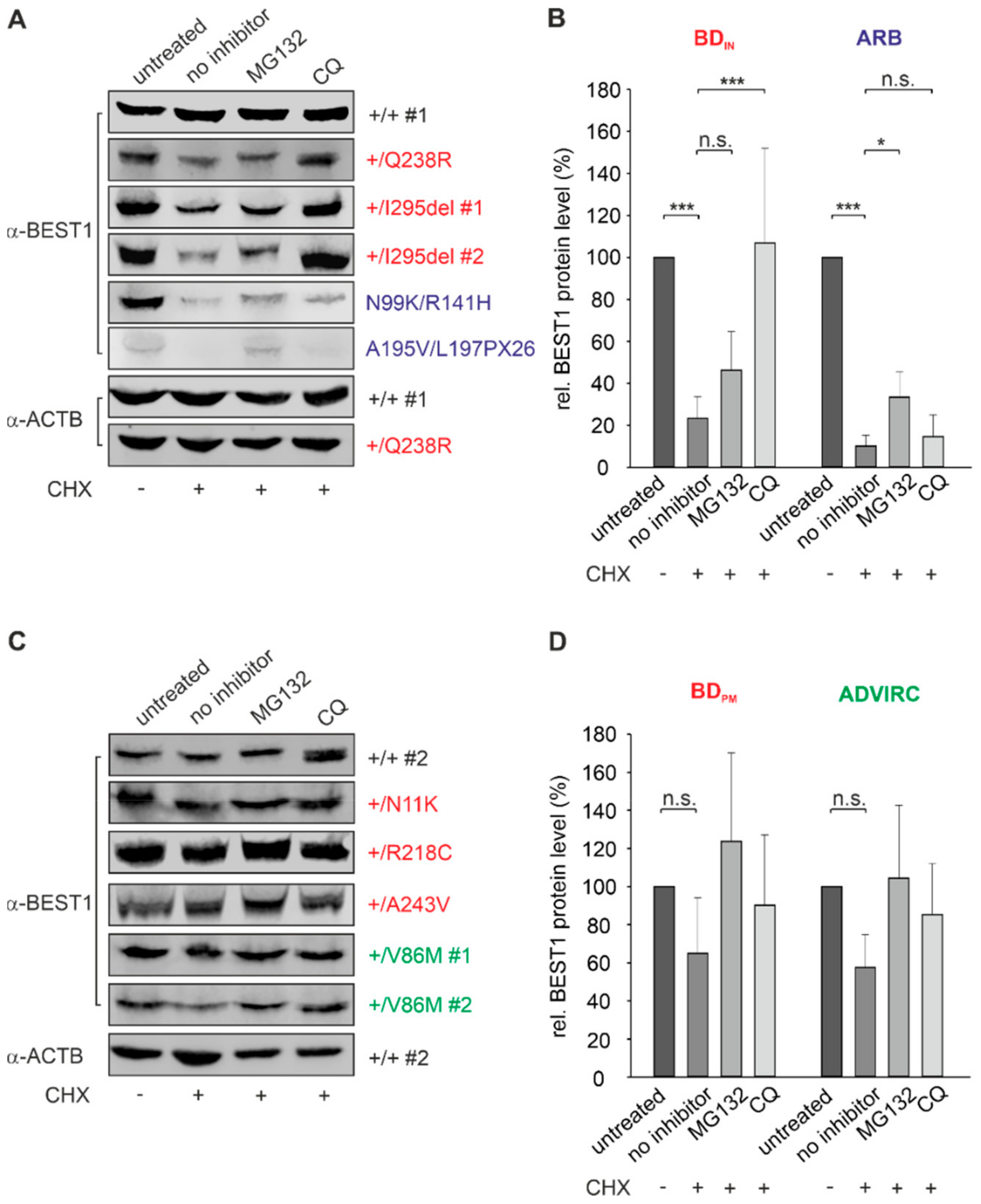
2.5. BEST1 Processing in BD and ARB hiPSC-RPEs via Distinct Degradation Pathways
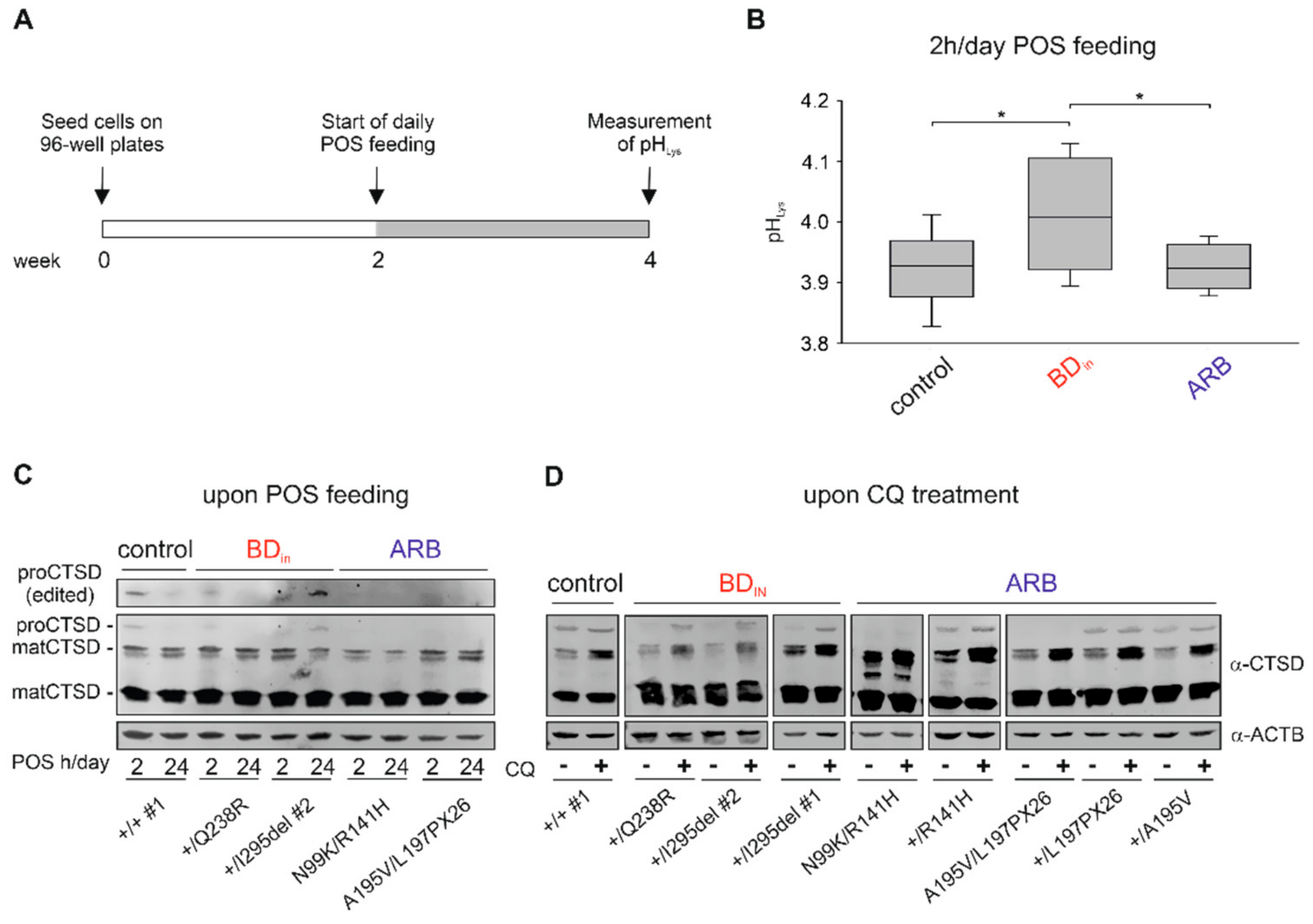
2.6. Altered Lysosomal Homeostasis in BD hiPSC-RPEs
2.7. Classification of BEST1 Mutations
3. Discussion
4. Materials and Methods
3D Mutation Modelling of BEST1 Variants
Generation of Human Induced Pluripotent Stem Cells (hiPSCs) and Differentiation into Retinal Pigment Epithelium (RPE) Cells
Measurement of Transepithelial Resistance (TER)
Antibodies
Immunofluorescence
RNA Isolation, Reverse Transcription (RT) and Real-Time Quantitative Reverse Transcriptase-Polymerase Chain Reaction (PCR) (qRT-PCR)
DNA Extraction, PCR Amplification and Sanger Sequencing
Cycloheximide (CHX) Treatment
Protein Sample Preparation, Sodium Dodecyl Sulfate (SDS) Page and Quantitative Western Blot Analysis
YFP-Halide Transport Assay
Isolation and Feeding of Photoreceptor Outer Segments (POS)
Lysosomal pH Measurement
Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tsunenari, T.; Sun, H.; Williams, J.; Cahill, H.; Smallwood, P.; Yau, K.W.; Nathans, J. Structure-function analysis of the bestrophin family of anion channels. J. Biol. Chem. 2003, 278, 41114–41125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartzell, H.C.; Qu, Z.; Yu, K.; Xiao, Q.; Chien, L.T. Molecular physiology of bestrophins: Multifunctional membrane proteins linked to best disease and other retinopathies. Physiol. Rev. 2008, 88, 639–672. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Prussia, A.; Yu, K.; Cui, Y.Y.; Hartzell, H.C. Regulation of bestrophin Cl channels by calcium: Role of the C terminus. J. Gen. Physiol. 2008, 132, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [Green Version]
- Brandl, C.; Zimmermann, S.J.; Milenkovic, V.M.; Rosendahl, S.M.; Grassmann, F.; Milenkovic, A.; Hehr, U.; Federlin, M.; Wetzel, C.H.; Helbig, H.; et al. In-Depth Characterisation of Retinal Pigment Epithelium (RPE) Cells Derived from Human Induced Pluripotent Stem Cells (hiPSC). Neuromol. Med. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, A.; Stohr, H.; Passmore, L.A.; Kramer, F.; Rivera, A.; Weber, B.H. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum. Mol. Genet. 1998, 7, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Petrukhin, K.; Koisti, M.J.; Bakall, B.; Li, W.; Xie, G.; Marknell, T.; Sandgren, O.; Forsman, K.; Holmgren, G.; Andreasson, S.; et al. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998, 19, 241–247. [Google Scholar] [CrossRef]
- Yardley, J.; Leroy, B.P.; Hart-Holden, N.; Lafaut, B.A.; Loeys, B.; Messiaen, L.M.; Perveen, R.; Reddy, M.A.; Bhattacharya, S.S.; Traboulsi, E.; et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Investig. Ophthalmol. Vis. Sci. 2004, 45, 3683–3689. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, G.M.; Kakuk, L.E.; Griesinger, I.B.; Simpson, S.A.; Nowak, N.J.; Small, K.W.; Maumenee, I.H.; Rosenfeld, P.J.; Sieving, P.A.; Shows, T.B.; et al. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics 1999, 58, 98–101. [Google Scholar] [CrossRef]
- Cross, H.E.; Bard, L. Electro-oculography in Best’s macular dystrophy. Am. J. Ophthalmol. 1974, 77, 46–50. [Google Scholar] [CrossRef]
- Nordstrom, S. Hereditary macular degeneration--a population survey in the country of Vsterbotten, Sweden. Hereditas 1974, 78, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Bitner, H.; Schatz, P.; Mizrahi-Meissonnier, L.; Sharon, D.; Rosenberg, T. Frequency, genotype, and clinical spectrum of best vitelliform macular dystrophy: Data from a national center in Denmark. Am. J. Ophthalmol. 2012, 154, 403–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingeist, T.A.; Kobrin, J.L.; Watzke, R.C. Histopathology of Best’s macular dystrophy. Arch. Ophthalmol. 1982, 100, 1108–1114. [Google Scholar] [CrossRef]
- Vincent, A.; McAlister, C.; Vandenhoven, C.; Heon, E. BEST1-related autosomal dominant vitreoretinochoroidopathy: A degenerative disease with a range of developmental ocular anomalies. Eye 2011, 25, 113–118. [Google Scholar] [CrossRef]
- Bitner, H.; Mizrahi-Meissonnier, L.; Griefner, G.; Erdinest, I.; Sharon, D.; Banin, E. A homozygous frameshift mutation in BEST1 causes the classical form of Best disease in an autosomal recessive mode. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5332–5338. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.L.; Hou, P.; Choy, K.W.; Chiang, S.W.; Tam, P.O.; Li, H.; Chan, W.M.; Lam, D.S.; Pang, C.P.; Lai, T.Y. Novel and homozygous BEST1 mutations in Chinese patients with Best vitelliform macular dystrophy. Retina 2010, 30, 820–827. [Google Scholar] [CrossRef]
- Dickson, V.K.; Pedi, L.; Long, S.B. Structure and insights into the function of a Ca (2+)-activated Cl (-) channel. Nature 2014, 516, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Liu, Q.; Kloss, B.; Bruni, R.; Kalathur, R.C.; Guo, Y.; Kloppmann, E.; Rost, B.; Colecraft, H.M.; Hendrickson, W.A. Structure and selectivity in bestrophin ion channels. Science 2014, 346, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.N.; Vaisey, G.; Long, S.B. Molecular mechanisms of gating in the calcium-activated chloride channel bestrophin. eLife 2019, 8. [Google Scholar] [CrossRef]
- Johnson, A.A.; Lee, Y.S.; Chadburn, A.J.; Tammaro, P.; Manson, F.D.; Marmorstein, L.Y.; Marmorstein, A.D. Disease-causing mutations associated with four bestrophinopathies exhibit disparate effects on the localization, but not the oligomerization, of Bestrophin-1. Exp. Eye Res. 2014, 121, 74–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, A.; Brandl, C.; Milenkovic, V.M.; Jendryke, T.; Sirianant, L.; Wanitchakool, P.; Zimmermann, S.; Reiff, C.M.; Horling, F.; Schrewe, H.; et al. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2630–E2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, V.M.; Rohrl, E.; Weber, B.H.; Strauss, O. Disease-associated missense mutations in bestrophin-1 affect cellular trafficking and anion conductance. J. Cell Sci. 2011, 124, 2988–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, A.; Milenkovic, V.M.; Wetzel, C.H.; Weber, B.H.F. BEST1 protein stability and degradation pathways differ between autosomal dominant Best disease and autosomal recessive bestrophinopathy accounting for the distinct retinal phenotypes. Hum. Mol. Genet. 2018, 27, 1630–1641. [Google Scholar] [CrossRef]
- Uggenti, C.; Briant, K.; Streit, A.K.; Thomson, S.; Koay, Y.H.; Baines, R.A.; Swanton, E.; Manson, F.D. Restoration of mutant bestrophin-1 expression, localisation and function in a polarised epithelial cell model. Dis. Models Mech. 2016, 9, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, A.D.; Johnson, A.A.; Bachman, L.A.; Andrews-Pfannkoch, C.; Knudsen, T.; Gilles, B.J.; Hill, M.; Gandhi, J.K.; Marmorstein, L.Y.; Pulido, J.S. Mutant Best1 Expression and Impaired Phagocytosis in an iPSC Model of Autosomal Recessive Bestrophinopathy. Sci. Rep. 2018, 8, 4487. [Google Scholar] [CrossRef]
- Johnson, A.A.; Lee, Y.S.; Stanton, J.B.; Yu, K.; Hartzell, C.H.; Marmorstein, L.Y.; Marmorstein, A.D. Differential effects of Best disease causing missense mutations on bestrophin-1 trafficking. Hum. Mol. Genet. 2013, 22, 4688–4697. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef]
- Moshfegh, Y.; Velez, G.; Li, Y.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. BESTROPHIN1 mutations cause defective chloride conductance in patient stem cell-derived RPE. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.; Kittredge, A.; Hopiavuori, A.; Ward, N.; Chen, S.; Fukuda, Y.; Zhang, Y.; Yang, T. Dual Ca(2+)-dependent gates in human Bestrophin1 underlie disease-causing mechanisms of gain-of-function mutations. Commun. Biol. 2019, 2, 240. [Google Scholar] [CrossRef]
- Sun, H.; Tsunenari, T.; Yau, K.W.; Nathans, J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA 2002, 99, 4008–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Kuai, D.; Guziewicz, K.E.; Meyer, J.; Wilson, M.; Lu, J.; Smith, M.; Clark, E.; Verhoeven, A.; Aguirre, G.D.; et al. Pharmacological Modulation of Photoreceptor Outer Segment Degradation in a Human iPS Cell Model of Inherited Macular Degeneration. Mol. Ther. J. Am. Soc. Gene Ther. 2015. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Shen, W.; Kuai, D.; Martin, J.M.; Guo, X.; Smith, M.A.; Perez, E.T.; Phillips, M.J.; Simonett, J.M.; Wallace, K.A.; et al. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum. Mol. Genet. 2013, 22, 593–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boon, C.J.; Theelen, T.; Hoefsloot, E.H.; van Schooneveld, M.J.; Keunen, J.E.; Cremers, F.P.; Klevering, B.J.; Hoyng, C.B. Clinical and molecular genetic analysis of best vitelliform macular dystrophy. Retina 2009, 29, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Carter, D.A.; Smart, M.J.; Letton, W.V.; Ramsden, C.M.; Nommiste, B.; Chen, L.L.; Fynes, K.; Muthiah, M.N.; Goh, P.; Lane, A.; et al. Mislocalisation of BEST1 in iPSC-derived retinal pigment epithelial cells from a family with autosomal dominant vitreoretinochoroidopathy (ADVIRC). Sci. Rep. 2016, 6, 33792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galietta, L.V.; Jayaraman, S.; Verkman, A.S. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am. J. Physiol. Cell Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef]
- Jayaraman, S.; Teitler, L.; Skalski, B.; Verkman, A.S. Long-wavelength iodide-sensitive fluorescent indicators for measurement of functional CFTR expression in cells. Am. J. Physiol. 1999, 277, C1008–C1018. [Google Scholar] [CrossRef]
- Davidson, A.E.; Millar, I.D.; Burgess-Mullan, R.; Maher, G.J.; Urquhart, J.E.; Brown, P.D.; Black, G.C.; Manson, F.D. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3730–3736. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, S.J.; Terashima, M.; Mizunuma, N.; Slapak, C.A. Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells: Participation of lysosomes. Blood 1997, 89, 3745–3754. [Google Scholar] [CrossRef] [Green Version]
- DePedro, H.M.; Urayama, P. Using LysoSensor Yellow/Blue DND-160 to sense acidic pH under high hydrostatic pressures. Anal. Biochem. 2009, 384, 359–361. [Google Scholar] [CrossRef]
- Liu, J.; Lu, W.; Reigada, D.; Nguyen, J.; Laties, A.M.; Mitchell, C.H. Restoration of lysosomal pH in RPE cells from cultured human and ABCA4(-/-) mice: Pharmacologic approaches and functional recovery. Investig. Ophthalmol. Vis. Sci. 2008, 49, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, B.; Dolenc, I.; Lenarcic, B.; Krizaj, I.; Turk, V.; Bieth, J.G.; Bjork, I. Acidic pH as a physiological regulator of human cathepsin L activity. Eur. J. Biochem. 1999, 259, 926–932. [Google Scholar] [CrossRef]
- Gieselmann, V.; Hasilik, A.; von Figura, K. Processing of human cathepsin D in lysosomes in vitro. J. Biol. Chem. 1985, 260, 3215–3220. [Google Scholar] [PubMed]
- Varghese, B.; Barriere, H.; Carbone, C.J.; Banerjee, A.; Swaminathan, G.; Plotnikov, A.; Xu, P.; Peng, J.; Goffin, V.; Lukacs, G.L.; et al. Polyubiquitination of prolactin receptor stimulates its internalization, postinternalization sorting, and degradation via the lysosomal pathway. Mol. Cell. Biol. 2008, 28, 5275–5287. [Google Scholar] [CrossRef] [Green Version]
- Duncan, L.M.; Piper, S.; Dodd, R.B.; Saville, M.K.; Sanderson, C.M.; Luzio, J.P.; Lehner, P.J. Lysine-63-linked ubiquitination is required for endolysosomal degradation of class I molecules. EMBO J. 2006, 25, 1635–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, R.; MacLaren, R.E.; Davidson, A.E.; Urquhart, J.E.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Keefe, R.O.; Black, G.C.; Manson, F.D. ADVIRC is caused by distinct mutations in BEST1 that alter pre-mRNA splicing. J. Med. Genet. 2009, 46, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Kaufman, S.; Packo, K.; Stohr, H.; Weber, B.H.; Goldberg, M.F. Long-Term Macular Changes in the First Proband of Autosomal Dominant Vitreoretinochoroidopathy (ADVIRC) Due to a Newly Identified Mutation in BEST1. Ophthalmic Genet. 2016, 37, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.Y.; Zhang, F.F.; Xu, J.; Wang, R.; Chen, J.; Logothetis, D.E.; Zhang, Z. Epilepsy-Related Slack Channel Mutants Lead to Channel Over-Activity by Two Different Mechanisms. Cell Rep. 2016, 14, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Guziewicz, K.E.; Cideciyan, A.V.; Beltran, W.A.; Komaromy, A.M.; Dufour, V.L.; Swider, M.; Iwabe, S.; Sumaroka, A.; Kendrick, B.T.; Ruthel, G.; et al. BEST1 gene therapy corrects a diffuse retina-wide microdetachment modulated by light exposure. Proc. Natl. Acad. Sci. USA 2018, 115, E2839–E2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdan, N.; Kritsiligkou, P.; Grant, C.M. ER stress causes widespread protein aggregation and prion formation. J. Cell Biol. 2017, 216, 2295–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.T.; Wu, W.H.; Lee, T.T.; Wu, W.P.; Xu, C.L.; Park, K.S.; Cui, X.; Justus, S.; Lin, C.S.; Jauregui, R.; et al. Clustered Regularly Interspaced Short Palindromic Repeats-Based Genome Surgery for the Treatment of Autosomal Dominant Retinitis Pigmentosa. Ophthalmology 2018, 125, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, J.H.; Jun, S.; Lee, J.H.; Bang, D. Selective targeting of KRAS oncogenic alleles by CRISPR/Cas9 inhibits proliferation of cancer cells. Sci. Rep. 2018, 8, 11879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Makiyama, T.; Harita, T.; Sasaki, K.; Wuriyanghai, Y.; Hayano, M.; Nishiuchi, S.; Kohjitani, H.; Hirose, S.; Chen, J.; et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 2017, 26, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, S.G.; Luoni, M.; Castoldi, V.; Massimino, L.; Cabassi, T.; Angeloni, D.; Demontis, G.C.; Leocani, L.; Andreazzoli, M.; Broccoli, V. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018, 27, 761–779. [Google Scholar] [CrossRef]
- Gao, X.; Tao, Y.; Lamas, V.; Huang, M.; Yeh, W.H.; Pan, B.; Hu, Y.J.; Hu, J.H.; Thompson, D.B.; Shu, Y.; et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 2018, 553, 217–221. [Google Scholar] [CrossRef]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef]
- Laird, P.W.; Zijderveld, A.; Linders, K.; Rudnicki, M.A.; Jaenisch, R.; Berns, A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991, 19, 4293. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid Exchange | Nucleotide Exchange | Alias (Clone Number) | Gender | Clinical Findings |
|---|---|---|---|---|
| +/+ #1 | - | MK (#22, #27b) | m | reference |
| +/+ #2 | - | AM (#13, #260) | f | reference |
| +/+ #3 | - | NG (#1) | f | reference |
| +/N11K * | 33T>G | MW (#232) | m | BD |
| +/R218C * | 652C>T | MO (#214) | m | BD |
| +/A243V | 728C>T | SK (#16) | m | BD |
| +/Q238R | 713A>G | DK (#Pka, #Pkb) | m | BD |
| +/I295del #1 * | 884_886delTCA | AP (#187) | m | BD |
| +/I295del #2 * | 884_886delTCA | MD (#18) | m | BD |
| N99K/R141H * | 422G>A/297C>A | LA (#29, #33) | f | ARB |
| +/N99K | 422G>A | EA (-) | f | ARB parent (healthy) |
| +/R141H * | 297C>A | MA (#74, #77) | m | ARB parent (healthy) |
| A195V/L197PX26 * | 584C>T/590_615del26 | TT (#178, #180) | m | ARB |
| +/A195V * | 584C>T | PT (#172, #176) | m | ARB parent (healthy) |
| +/L197PX26 * | 590_615del26 | AT (#166, #168) | f | ARB parent (healthy) |
| +/V86M #1 * | 256G>A | IS (#53a, #53b) | f | ADVIRC |
| +/V86M #2 * | 256G>A | JS (#52a, #52b) | m | ADVIRC |
| Class I |
|
| Class II |
|
| Class III |
|
| Class IV |
|
| Class V |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nachtigal, A.-L.; Milenkovic, A.; Brandl, C.; Schulz, H.L.; Duerr, L.M.J.; Lang, G.E.; Reiff, C.; Herrmann, P.; Kellner, U.; Weber, B.H.F. Mutation-Dependent Pathomechanisms Determine the Phenotype in the Bestrophinopathies. Int. J. Mol. Sci. 2020, 21, 1597. https://doi.org/10.3390/ijms21051597
Nachtigal A-L, Milenkovic A, Brandl C, Schulz HL, Duerr LMJ, Lang GE, Reiff C, Herrmann P, Kellner U, Weber BHF. Mutation-Dependent Pathomechanisms Determine the Phenotype in the Bestrophinopathies. International Journal of Molecular Sciences. 2020; 21(5):1597. https://doi.org/10.3390/ijms21051597
Chicago/Turabian StyleNachtigal, Anna-Lena, Andrea Milenkovic, Caroline Brandl, Heidi L. Schulz, Lisa M. J. Duerr, Gabriele E. Lang, Charlotte Reiff, Philipp Herrmann, Ulrich Kellner, and Bernhard H.F. Weber. 2020. "Mutation-Dependent Pathomechanisms Determine the Phenotype in the Bestrophinopathies" International Journal of Molecular Sciences 21, no. 5: 1597. https://doi.org/10.3390/ijms21051597