Article Text

Abstract
Age-related macular degeneration (AMD) represents a leading cause of blindness worldwide. Neovascular AMD (nAMD) is a subtype of AMD most frequently treated with intravitreal anti-vascular endothelial growth factor (aVEGF) injections, which has allowed for patients to maintain vision that would have otherwise been lost. However, the need for frequent intravitreal injections for optimal results poses a risk for undertreatment in nAMD patients due to the high treatment burden associated with current aVEGF therapy. Many novel agents and pathways are being explored and targeted for less burdensome treatment options, one of which is the ranibizumab port delivery system (PDS). The PDS is a surgically implanted, refillable device that allows for the sustained release of ranibizumab, a widely used aVEGF agent, into the vitreous cavity. Positive results non-inferior to monthly ranibizumab injections in both phase II and phase III clinical trials allowed for FDA approval of the device with refill intervals of 6 months, which represents the longest approved treatment interval to date for nAMD therapy. This article reviews the need for a durable nAMD treatment option in real-world practice, the clinical trial and extension study data for the PDS, the risk of adverse events and safety profile of the PDS and the potential clinical role of the PDS in answering the real-world needs of nAMD treatment. In addition, other pipeline sustained-treatment modalities are discussed in the context of ongoing clinical trials.
- Retina
- Angiogenesis
- Macula
- Treatment Surgery
- Neovascularisation
- Degeneration
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Age-related macular degeneration (AMD) remains one of the leading causes of vision loss worldwide; it is estimated to account for 8.7% of global blindness in those that are 45–85 years old.1 As the world’s ageing population continues to grow, it is predicted that AMD will become increasingly prevalent, with estimates of global AMD cases potentially reaching as high as 288 million by 2040.1
The pathology of AMD can be grossly divided into two main categories: dry or non-neovascular AMD (nAMD), and wet or nAMD. Although dry AMD accounts for nearly 90% of all cases of AMD, it is nAMD that is primarily responsible for rapid and severe loss of vision.2 3 nAMD is characterised by the proliferation and leakage of new vasculature within the retina, subretinal space and/or under the retinal pigment epithelium (RPE).3–5 The associated drusen deposits seen in AMD in combination with inflammation and activation of the complement cascade are thought to reduce nutrient flow and/or waste removal from the RPE and Bruch membrane via the choriocapillaris, which could promote further neovascularisation.3–5 Compared with normal vasculature, the abnormal blood vessels in nAMD are prone to exudation and haemorrhage, which can acutely, and dramatically, decrease visual acuity. These processes can ultimately lead to photoreceptor death and irreversible vision loss through both apoptosis and necroptosis.3–5
The expression of vascular endothelial growth factor (VEGF), a proangiogenic cytokine, was found to be a leading factor in the development of neovascularisation and increased vascular permeability seen in nAMD.3 6 7 Since this discovery, the refined roles of VEGF and other signalling molecules have been further elucidated and include the regulation of vasculogenesis, angiogenesis, lymphangiogenesis, vascular permeability and endothelial cell proliferation and migration.3 8 9 The VEGF family of molecules includes VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor, with numerous subtypes created via alternative mRNA splicing. Of these, VEGF-A represents the current primary target in nAMD treatment.8 VEGF-A binds to the extracellular domains of tyrosine kinase receptors VEGR-1 and VEGR-2, leading to downstream cellular signalling that promotes angiogenesis and vascular permeability.9 10 Alternatively, VEGF-C/D acts primarily on VEGFR-3 and are thought to primarily regulate lymphangiogenesis while assisting in regulating angiogenesis in part through a weak affinity for VEGFR-2.9
History of anti-VEGF therapy
The introduction of pegaptanib sodium in 2004 marked the first anti-VEGF (aVEGF) intravitreal injection approved by the FDA.11 Although the usage of pegaptanib has decreased in favour of more efficacious aVEGF injections such as, aflibercept, brolucizumab, ranibizumab and bevacizumab (off-label), it marked a fundamental shift in the treatment paradigm of nAMD.12 Current treatment options all act to inhibit neovascularisation and vascular permeability by binding specific molecules within the VEGF family or by binding to and mimicking specific VEGF receptors. Aflibercept is a receptor decoy created by fusing the Fc portion of human IgG1 to the VEGFR-1 and VEGFR-2 binding domains and binds VEGF-A, VEGF-B and placental growth factor.10 12 Brolucizumab is a single-chain antibody fragment that binds to and inhibits all isoforms of VEGF-A with high affinity.10 12 In contrast, ranibizumab and bevacizumab are IgG1 monoclonal antibodies that target VEGF-A and prevents it from binding its respective tyrosine kinase receptor, thus inhibiting angiogenesis and increased vascular permeability.10 12
In 2006, the MARINA and ANCHOR registration trials demonstrated that monthly ranibizumab injection led to significantly less visual acuity decline and aggregate visual improvement in comparison with sham and with photodynamic therapy. These findings led to the FDA approval of ranibizumab for nAMD in 2006 and its subsequent establishment as standard of care.13 14
Disparities between clinical trials and clinical practice
Although frequent ranibizumab injections have proven effective in halting the progression of vision loss in many patients with nAMD, the monthly injection schedule studied with the initial registration trials exemplifies a significant treatment burden for patients in addition to the increased cost and risk inherent to repeated intravitreal injections.15 In addition, observational studies in real-world clinical practice have shown that patients are unable to achieve similar benefits as those seen in clinical trials due to the high associated treatment burden.2 16 17 Since then, studies were initiated to determine treatment regimens that would form a compromise between patient burden and adequate treatment coverage. The ‘pro re nata’ (PRN) approach and ‘treat-and-extend’ (T&E) approach are two non-continuous dosing schedules that have been proposed as alternatives to continuous, monthly injections. The PRN regimen requires monthly monitoring visits to approach the effectiveness of monthly ranibizumab treatment and treating patients with aVEGF injections when disease activity recurs. In contrast, T&E involves injecting diseased eyes at every visit, but incrementally extending/reducing the treatment interval (frequently by 2 weeks intervals) depending on disease activity.6 7 18–20 The efficacy of T&E regimens was studied in the TREND study, which compared the T&E model against the standard monthly dosing schedule. This study was a 12-month, multicentre randomised controlled trial with 650 treatment-naive nAMD patients that assessed the efficacy and safety of ranibizumab 0.5 mg dosed via the T&E model in comparison to the established, monthly regimen.21 The trial concluded that ranibizumab dosed via the T&E model was statistically non-inferior (p<0.001) to monthly treatment when measuring end of study visual acuity and led to both fewer injections (8.7 vs 11.1) and clinic visits (8.9 vs 11.2) compared with the monthly regimen.
Between the possible regimens, studies have shown that proactive treatment and more frequent dosing leads to improved visual outcomes (fixed and T&E vs PRN).18 20 In addition, PRN dosing highlighted the dangers of undertreatment with patients achieving the worst VA outcomes of any regimen.6 19 20 Other studies have added that T&E can lead to non-inferior visual acuity outcomes in nAMD patients while reducing the burden on both the patient and healthcare system overall. For example, a meta-analysis that included 26 360 patients from 42 real-world studies involving the use of ranibizumab under T&E, T&E plus loading dose or PRN regimen concluded that patients who used solely a T&E model sustained high levels of visual acuity at the 1-year, 2-year and 3-year mark (+8.8, +6.7, +5.4) measured via Early Treatment Diabetic Retinopathy Study (ETDRS) letters.2 6 15 22 Thus, as T&E theoretically achieves non-inferior VA benefits vs standard monthly dosing in the TREND study and other above referenced studies while requiring fewer intravitreal injections and clinic visits in comparison to PRN, it can ease treatment burden for patients and providers, increase patient compliance and increase the patient volume providers can treat.6 19–21 These factors allowed T&E regimens to become the standard clinical practice of many providers.18 23–27
However, despite these promising results, other studies have observed inferior VA results in real-world T&E treatment regimens in comparison with the controlled treatment provided in clinical trials.17 24 25 One study found that only 13% of patients under T&E regimens were truly able to reach and maintain a 12-week treatment interval, only 10% able to maintain an 8-week interval, and 27% were not able to maintain even an 8-week interval.26 This disparity between real-world and clinical trial VA outcomes of T&E regimens is thought to be due to the still-relatively-high treatment burden of frequent office visits, which itself represents the summative difficulties of patient and provider time/scheduling constraints, the need for caregivers to accompany patients, socioeconomic factors, and dissatisfaction with initial visual acuity results.24 25 28 Given mixed results from real-world studies that use the T&E regimen, in addition to its risk of undertreating patients highlighted in some studies, a need remains for alternative, longer-lasting treatments that are comparable in efficacy to the original standard set by monthly ranibizumab in its pivotal clinical trials while foregoing its burdensome administration schedule.17 24 25
The port delivery system: a promising tool
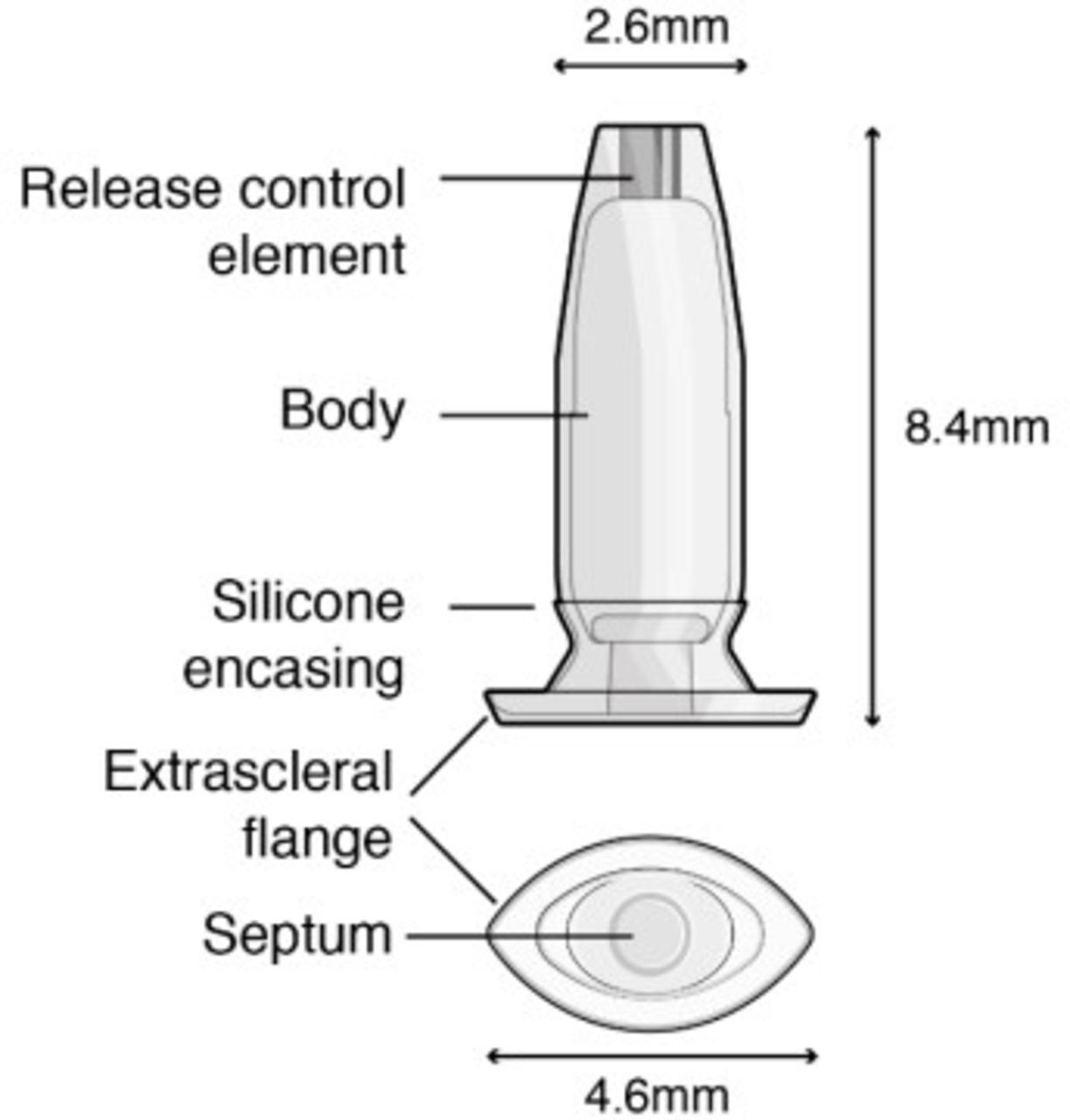
The development and recent FDA approval of the ranibizumab port delivery system (PDS) (Susvimo, Genentech), a permanent, surgically implanted, refillable device, is a potential answer to current clinical practice needs.29–32 The PDS is a hollow drug reservoir composed of a nonbiodegradable polysulfone body coated in silicone, capable of holding 0.02 mL of drug, which is inserted into the pars plana through a 3.5 mm scleral incision and anchored within the sclera by an extrascleral flange (figure 1). The implant measures 2.6 mm at its tip, 8.4 mm in length, and 4.6 mm at the flange. The PDS allows for the sustained delivery of ranibizumab through the micro fenestrated titanium release control element. Drug delivery is achieved via passive diffusion into the vitreous cavity using Fick’s law of diffusion along a concentration gradient between the high concentration of ranibizumab in the implant and the lower concentration in the vitreous. The drug inside the PDS can undergo refill-exchange in the clinic through a self-sealing, silicone septum and is refilled with a proprietary dual-bore needle that allows for the simultaneous withdrawal of leftover ranibizumab within the reservoir while injecting 0.1 mL of new ranibizumab solution.29–32 The new ranibizumab solution is injected at a volume five times greater (0.1 mL) than the volume of the reservoir (0.02 mL) to achieve a refill-exchange efficiency greater than 98%.33 The phase II LADDER trial was a randomised, multicentre, active-treatment controlled trial that involved 220 nAMD patients who had been treated with and were responsive to at least two aVEGF injections. Patients were randomised 3:3:3:2 for treatment with the PDS with concentrations of ranibizumab at 10 mg/mL, 40 mg/mL, 100 mg/mL or monthly 0.5 mg ranibizumab intravitreal injection.31 34 Although the study was temporarily paused due to frequent vitreous haemorrhages (VH), an alteration to the surgical technique—laser cauterising the choroid before making the incision for the implant—allowed for safer implantation (5% vs the initial 50% VH rate) and continuation of the clinical trial. At month 22 of the LADDER trial, mean change in best-corrected visual aAcuity (BCVA) from baseline was ‒4.6 ETDRS letters, ‒2.3 ETDRS letters, +2.9 ETDRS letters and +2.7 ETDRS letters in patients treated with PDS 10 mg/mL, 40 mg/mL, 100 mg/mL and monthly intravitreal 0.5 mg ranibizumab injection, respectively. The primary endpoint was median time to first refill of the PDS and was measured as 8.7 months, 13.0 months and 15.8 months, in the 10 mg/mL, 40 mg/mL and 100 mg/mL PDS arms, implying a high potential for reduced overall treatment burden and protection against undertreatment secondary to missed appointments. The LADDER trial assessed mean change in central foveal thickness (CFT) of the retina and observed similar mean reductions in CFT between the patients in the 100 mg/mL PDS arm and in the monthly 0.5 mg ranibizumab injection arm (‒4.0 µm, and ‒10.9 µm). The LADDER study concluded that PDS 100 mg/mL was comparable to standard 0.5 mg ranibizumab injections over the course of the 22-month study and generated the rationale for the phase III programme.31
{kind=link}
Diagram of the ranibizumab port delivery system detailing components and dimensions measured in millimetres.
The comparable efficacy of 100 mg/mL PDS to monthly 0.5 mg ranibizumab injection in addition to the potential for reduced treatment burden via the observed median refill time of 15.8 months promoted its further study in Genentech’s phase III ARCHWAY trial.30 The ARCHWAY trial (NCT03683251) was an open-label, randomised, multicentre, active-comparator trial in which 418 patients who had received and were responsive to, at least three aVEGF injections were randomly assigned 3:2 to two arms: treated with 100 mg/mL PDS refilled at fixed, 24-week intervals, or monthly 0.5 mg ranibizumab intravitreal injections.30 ARCHWAY’s primary outcome was to assess mean change in BCVA and central subfield thickness (CST) of the retina in both treatment arms at week 40 as compared with baseline. ARCHWAY concluded that the 100 mg PDS refilled every 24 weeks was noninferior to monthly 0.5 mg ranibizumab injections, with mean change in BCVA averaged over weeks 36–40 reported as +0.2 ETDRS letters and +0.5 ETDRS letters in the PDS and monthly injection arms, respectively.30 Following successful phase III trial results, the ranibizumab PDS was approved by the Food and Drug Administration in October of 2021.
An ongoing extension study, PORTAL, was initiated for patients in either treatment arm who had participated in either the phase II LADDER study or phase III ARCHWAY study.35 These patients were those who have rolled over from the LADDER trial, the 24-week regimen of the ARCHWAY study, or for patients from the monthly injection arms who had been newly implanted with the PDS at entry into PORTAL. The PORTAL extension study is ongoing and evaluates the long-term safety and tolerability of the ranibizumab PDS 100 mg/mL given at 24-week intervals across the study duration of 240 weeks. The study’s primary outcome is to assess for any ocular or systemic adverse events, while the study’s secondary outcome is to measure mean changes in BCVA and in CST from baseline over 240 weeks. The PORTAL study is estimated to be completed in 2026.
Risks and benefits of the PDS
Although the ranibizumab PDS shows promise in reducing nAMD treatment burden, the ranibizumab PDS does carry risk. As seen in some patients within the phase II and phase III clinical trials, adverse events inherent to ocular surgery including infection, eye pain, VH and retinal detachment may occur.29 30 32 Notably, the US label for the PDS includes a black-box warning for endophthalmitis, which states that the implant has been associated with a threefold higher rate of endophthalmitis compared with monthly intravitreal injections of ranibizumab. In the phase II and III clinical trials, 2% of patients receiving an implant experienced an episode of endophthalmitis.36 In ARCHWAY, four patients who received the PDS experienced endophthalmitis, and two required removal of the PDS.30 In LADDER, three PDS patients experienced endophthalmitis and all three required discontinuation of treatment and explantation of the PDS.30 31 The majority of these endophthalmitis events have been associated with exposure of the implant via either conjunctival erosion or retraction.37 The label also states 3.6% of patients receiving a PDS implant experience conjunctival erosion and 1.6% of patients experience conjunctival retraction in clinical trials.36 Other noted risks include rhegmatogenous retinal detachment, implant dislocation, conjunctival blebs and transient postoperative decreases in visual acuity, as expected with intraocular surgery. In aggregate, vision returned to baseline within 2 months of implantation.33 Implant dislocation occurred in one patient in ARCHWAY following a refill-exchange procedure and was thought to be related to a scleral incision greater than the specified range for PDS implantation (3.7 vs 3.5 mm).30 Phakic and pseudophakic eyes were included across treatment arms, with around 40% of patients characterised as phakic and 60% as pseudophakic in the ARCHWAY trial.30 Through week 40, equivalent proportions (9.5%) of phakic patients in both the PDS and monthly ranibizumab arms experienced new or worsening cataracts, highlighting no PDS-associated risk of cataract development in clinical trials.30 Still, the US label notes a possibility for traumatic cataract development if the PDS is implanted incorrectly, allowing for contact between the PDS and the lens of the eye.36 There were also no cases of raised intraocular pressure associated with the PDS in clinical trials30 31 34 However, hypotony was observed in 6% of patients who received the PDS in clinical trials compared with 0% in patients receiving monthly ranibizumab injections.30 36 The most frequent serious ocular adverse event experienced during clinical trials was VH. As previously mentioned, the surgical technique was amended following an initial high rate of VHs during the phase 2 LADDER study.35 Prior to surgical technique optimisation, the rate of VH was 50% for the initial 22 patients treated with the PDS. However, this number was reduced to 5.1% (8/157 patients) overall following the laser choroid cauterisation amendment.31 During the phase III, ARCHWAY study, 13 patients (5.2% overall) experienced VH from the PDS arm.30 As clinicians become more experienced with the procedure and as techniques continue to improve, it is expected that complication rates may further reduce. There is ongoing concern about the potential association of geographic atrophy (GA) with VEGF suppression in nAMD. This highlights a potential risk of the patients treated with PDS in that its longer-term aVEGF activity could result in increasing incidences of GA.38
Despite these risks, the ranibizumab PDS has the potential to maintain results comparable to monthly ranibizumab treatment while reducing treatment burden by extending the treatment interval to a biannual basis, or potentially even longer, as seen by the extended median time to first refill in the Phase II LADDER clinical trial (15.8 months with 100 mg/mL PDS).31 This 6-month interval represents the longest of any currently approved therapies for nAMD, and the associated reduction in treatment burden could help alleviate many of the factors that lead to undertreatment in nAMD patients, including time constraints, caregiver burden and discomfort and risks associated with repeated intravitreal injection.24 25 28 Results from the phase III ARCHWAY trial support this; 218 of 234 PDS-treated patients (93.2%) stated they preferred the PDS over intravitreal injections at the end of the study.28 29 A questionnaire provided to a subset of patients found that their top reasons for preferring the PDS included fewer treatments and less discomfort, which support the goal of the PDS to reduce treatment burden and increase compliance for patients.37 In addition, due to the continuous aVEGF delivery provided by the PDS, visual acuity results similar to those seen in clinical trials may be more attainable in clinical practice compared with the respective differences observed when using traditional intravitreal injections in practice vs in clinical trials. Finally, the selection of ranibizumab as the deliverable agent may optimise the PDS’s effectiveness as well as adoption into clinical practice for nAMD as well as other diseases such as diabetic retinopathy and branch retinal vein occlusion as ranibizumab is a widely established, effective, and well-tolerated aVEGF drug already used to treat such patients.39 Alongside this, a study by Chandrasekaran et al supported the long-term biocompatibility of the PDS and its non-inflammatory and non-toxic nature.40
Other sustained release/long-duration treatments in development
Alongside the ranibizumab PDS, innovative drugs and novel treatment modalities in the pipeline aim to further prolong treatment intervals via novel delivery or by targeting novel pathways implicated in nAMD pathology, including tyrosine kinase inhibitors, aVEGF biopolymers, ANG-2 inhibitors, TIE-2 inhibitors, integrin inhibitors, and gene therapies.41 Alternative sustained-release treatments targeting the tyrosine kinase pathway currently in development include GB-102 (Sunitinib maleate; GrayBug Vision, Redwood City, California, USA) and Durasert Bioerodible TKI (Durasert; EyePoint Pharmaceuticals, Watertown, Massachusetts, USA).2 15 41
GB-102, or sunitinib, is a tyrosine kinase inhibitor (TKI) delivered via intravitreal injection and inhibits VEGF-R 1, 2 and 3, blocking VEGF-A,B,C,D, and placental growth factor.2 41 Sunitnib is contained inside biodegradable polymer nanoparticles that allow for sustained therapeutic action, requiring only biannual treatment.2 41 ALTISSIMO is a phase 2b multicentre, masked, randomised active-controlled study that aims to compare both GB-102 1 mg and GB-102 2 mg delivered every 6 months to intravitreal aflibercept injections every 2 months and used median time to first supportive therapy as its primary endpoint.42 Following safety concerns, the GB-102 2 mg arm was halted, and patients received GB-102 1 mg for their second dose. The median time to first supportive therapy for the GB-102 1 mg formulation was 5 months, with 62% of patients foregoing supportive therapy for 4 or more months.2 Mean change in BCVA was evaluated via ETDRS and found to be nine letters lower in the GB-102 1 mg group and was thought to be associated with preexisting uncontrollable disease and particle dispersion.2 Of note, this study used a new formulation in efforts to mitigate particle dispersion to the anterior chamber observed in the prior phase 1/2a ADAGIO trial.2 41 Despite this, 3 of 21 patients had transient GB-102 particles in the anterior chamber, and 4 of 21 patients experienced intraocular inflammation which required corticosteroid treatment as noted in recent extension data.43 Although GB-102’s pan-VEGF inhibition has potential to provide more comprehensive coverage in nAMD patients, its current issues involving intraocular inflammation and particle dispersion must be addressed before becoming a viable alternative to the ranibizumab PDS. Nonetheless, GB-102’s delivery via intravitreal injection is advantageous as it is less invasive than surgery and vitreoretinal surgeons are experienced in intravitreal injections. Finally, clinical trial data thus far observed a mean change of 9 ETDRS letters lower in patients receiving GB-102 1 mg as compared with aflibercept injections every 2 months.2 As additional trials with larger numbers of patients are completed, the efficacy of GB-102 can be further elucidated to determine its potential as an alternative, less invasive, sustained aVEGF treatment platform.
EYP-1901 uses a novel TKI, vorolanib, combined with a proprietary delivery platform that allows for implantation via intravitreal injection.2 41 EYP-1901 is currently undergoing a phase I, open-label, dose escalation study in 17 nAMD patients (DAVIO) that will evaluate safety as its primary endpoint and BCVA and CST as secondary endpoints.44 Interim results thus far are promising, with 76% of eyes requiring no supplementary aVEGF injections at 4 months, 53% of eyes at 6 months and 41% of eyes at 9 months.45 In addition, BCVA and CST remained stable at 8 months (−3 ETDRS letters and+13 µm, respectively).45 Similar to GB-102, EYP-1901’s mechanism of delivery is less invasive than surgical implantation of the PDS while providing sustained treatment to nAMD patients. The ranibizumab PDS, however, uses an already established and well-tolerated aVEGF drug in comparison to these two upcoming sustained aVEGF treatment modalities.
Conclusion
Until the efficiency and safety of various pipeline therapies can become competitive with existing treatment options, the ranibizumab PDS may establish itself as a practical option for providing sustained delivery of aVEGF agents to patients at a high risk of undertreatment, potentially reducing treatment burden while allowing physicians to care for greater patient volumes. The rate of endophthalmitis, a significant risk of the PDS, may decrease if conjunctival retraction and erosion can be mitigated, detected and managed promptly, or perhaps reduced in incidence if further changes in surgical technique are made. The introductory adoption of the PDS has helped pave the way for novel, sustained-release treatment delivery for retinal disease, and the possibility of employing either the PDS or other agents and delivery systems for common retinal diseases such as retinal vein occlusions and diabetic retinopathy. Ultimately, real-world data on the usage and efficacy of the ranibizumab PDS will help shape future treatment paradigms, as many clinicians look forward to offering patients additional, less burdensome treatment options as they become available.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors DAE conceived of this review concept. All authors contributed to the authorship and editing of this review.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests DAE is a consultant, speaker, and investigator for Genentech.
Provenance and peer review Not commissioned; internally peer reviewed.