Article Text

Abstract
Objective To evaluate factors associated with Diabetic Retinopathy Severity Scale (DRSS) changes with less frequent ranibizumab after induction therapy.
Methods and analysis Post hoc analyses of RIDE/RISE and their open-label extension (OLE). Analyses included patients with diabetic retinopathy (DR)/diabetic macular oedema who completed the OLE. Comparisons were made between patients with improved/maintained (≥0 step decrease from OLE baseline (month 36) to month 48) versus worsened (≥1 step increase) DRSS during the OLE. DRSS changes over 12 months were compared between patients randomised to ranibizumab at RIDE/RISE baseline who improved to DRSS score ≤43 at OLE baseline (induced) versus those randomised to sham with DRSS score ≤43 at RIDE/RISE baseline (native).
Results From OLE baseline to month 48, 72% (263/367) of patients improved/maintained DRSS scores. These patients had similar mean best-corrected visual acuity at RIDE/RISE (56.4 letters) and OLE baseline (68.6 letters) versus patients with worsened scores (58.2 and 70.8 letters). Patients who improved/maintained DRSS scores had similar mean central foveal thickness at RIDE/RISE (492 µm) and OLE baseline (196 µm) versus patients with worsened scores (441 and 167 µm). Patients who improved/maintained DRSS scores received a significantly higher (p<0.0001) mean number of pro re nata (PRN) injections (4.4) between OLE baseline and month 48 versus those with worsened scores (2.3). Patients with more severe DR at baseline who achieved mild-to-moderate non-proliferative DR (NPDR) induced by monthly ranibizumab injections were significantly more likely to worsen (p<0.0001) than those with mild-to-moderate NPDR at baseline randomised to sham injections (1.0-step versus 0.1-step worsening).
Conclusions Most patients improved/maintained DRSS scores with less-than-monthly PRN ranibizumab. Some minimum treatment/monitoring may be necessary to maintain improvements after induction therapy.
Trial registration numbers NCT00473382/NCT00473330.
- vision
- treatment medical
- retina
Data availability statement
Data are available upon reasonable request. For eligible studies qualified researchers may request access to individual patient level clinical data through a data request platform. At the time of writing this request platform is Vivli. https://vivli.org/ourmember/roche/. For up-to-date details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing. Anonymised records for individual patients across more than one data source external to Roche cannot, and should not, be linked due to a potential increase in risk of patient re-identification.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
Vascular endothelial growth factor (VEGF)-A is upregulated in patients with diabetic retinopathy (DR), and intravitreal injections of VEGF inhibitors have been shown to reduce DR severity. However, the durability of this improvement is not well understood.
What this study adds
This post hoc analysis (RIDE/RISE and their open-label extension (OLE)) showed that most ranibizumab-treated patients had improved/maintained DRSS with less-than-monthly pro re nata treatment. More severe DR at baseline was associated with greater clinical benefit from monthly ranibizumab treatment during RIDE/RISE, but was associated with Diabetic Retinopathy Severity Scale (DRSS) score instability with intermittent dosing in the OLE.
How this study might affect research, practice or policy
DRSS improvements with monthly therapy can be maintained with less-than-monthly treatment. Induced DRSS improvements appear more volatile than untreated DRSS of the same level.
Introduction
Diabetic retinopathy (DR) is the most common microvascular complication of diabetes. In 2010, more than 7 million Americans were estimated to have DR; this prevalence is projected to more than double to 15 million by 2050.1 Vascular endothelial growth factor (VEGF)-A is upregulated in patients with DR, and intravitreal injections of VEGF inhibitors have been shown to reduce DR severity.2 However, the durability of such improvement is not well understood. The RIDE/RISE trials of ranibizumab in patients with DR and diabetic macular oedema (DME), together with the open-label extension (OLE) study of these trials, offer up to 5 years of follow-up in patients receiving anti-VEGF therapy. Furthermore, there is a switch at year 3 from monthly treatments to a less-intensive pro re nata (PRN) regimen. This trial design offers a unique opportunity to investigate what happens with DR improvements achieved with intensive VEGF suppression once monthly treatment is reduced. Here, we report the findings of post hoc analyses of RIDE/RISE OLE data, which aim to characterise the changes in Diabetic Retinopathy Severity Scale (DRSS) score after induction therapy ends and identify factors that may be associated with changes in DRSS outcomes with intermittent ranibizumab treatment over time.
Methods
Study designs
Post hoc analyses were performed using data from the RIDE and RISE trials and the OLE. The study designs for these trials have been described in detail elsewhere.3–5 In RIDE/RISE, 759 patients with DME were randomised to receive monthly ranibizumab 0.3 mg or 0.5 mg or sham injections with rescue laser therapy (according to prespecified criteria, including central foveal thickness (CFT) of ≥250 µm with a <50-µm change from the previous month, and no previous macular laser in the previous 3 months) for 24 months.5 At month 25, patients in the sham arm crossed over to monthly ranibizumab 0.5 mg, whereas patients in the other ranibizumab arms continued their original ranibizumab regimen for the next 12 months.5 All patients who had not discontinued treatment and had completed month 36 of RIDE/RISE were eligible to enrol in the OLE, which continued for an additional 24 months or until 30 days after the US Food and Drug Administration approval of ranibizumab for the treatment of DME, if that occurred within 24 months of joining the OLE.3 6 Overall, 500/582 eligible patients chose to participate in the OLE; their demographic characteristics were well balanced between the three previous study treatment arms, as previously described.3
Patients in the OLE received ranibizumab 0.5 mg, administered PRN according to the following prespecified criteria: DME diagnosed by the investigator based on optical coherence tomography (OCT) or worsening in best-corrected visual acuity (BCVA) by at least five Early Treatment Diabetic Retinopathy Study letters from the start of the OLE at month 36.3 Re-treatment was not based on DR severity. DME was defined as the presence of intraretinal fluid or cysts, subretinal fluid or subretinal pigment epithelium fluid; no absolute macular or central subfield thickness criteria mandated treatment. After ranibizumab injection, patients were observed every 30±7 days, or every 60±7 days or every 90±7 days at investigator discretion.3 The month 48 visit was mandatory for all patients in the OLE, regardless of the injection interval duration, because this provided a 12-month OLE assessment time point.3
Patient and public involvement
Patients or the public were not involved in the design, conduct, reporting or the dissemination plans for RIDE/RISE.
Post hoc analyses
The following definitions were used for the post hoc analyses: ‘RIDE/RISE baseline’ was defined as the baseline visit for RIDE/RISE that occurred at month 0; ‘OLE baseline’ was defined as the ‘new’ baseline starting at the month 36 visit, when the RIDE/RISE core trials ended and the OLE trial began; ‘native’ mild-to-moderate non-proliferative DR (NPDR) was defined as patients randomised to sham injections who had a DRSS score ≤43 at RIDE/RISE baseline; ‘induced’ mild-to-moderate NPDR was defined as patients randomised to monthly ranibizumab treatment who had a DRSS score >43 (ie, moderate, moderately severe or severe NPDR; or proliferative DR (PDR)7) at RIDE/RISE baseline and experienced improvements to DRSS score ≤43 (mild-to-moderate NPDR) by OLE baseline.
We explored the relationships between (1) improved or maintained DRSS score versus worsened DRSS score during the OLE and study eye BCVA and CFT at RIDE/RISE baseline and OLE baseline; and (2) improved or maintained DRSS score versus worsened DRSS score during the OLE and the number of PRN injections administered between OLE baseline and month 48. Improved or maintained DRSS score was defined as a ≥0-step decrease from OLE baseline to month 48, whereas worsened DRSS score was defined as an at least one-step increase over the same time frame.
We also compared DR outcomes between the native and induced mild-to-moderate NPDR subgroups over 12 months of follow-up (from RIDE/RISE baseline to month 12 and from OLE baseline (month 36) to month 48, respectively; online supplemental figure S1). This included mean changes in DRSS score and individual DRSS score changes from baseline.
Supplemental material
Individual DRSS score changes were examined in more detail for the induced mild-to-moderate NPDR subgroup to (1) compare outcomes from RIDE/RISE baseline to month 36 with changes from OLE baseline to month 48 (including step changes in DRSS score, ie, movement from one category to another); (2) assess DRSS score changes in patients diagnosed with PDR at RIDE/RISE baseline; and (3) quantify DRSS score changes for patients without previous pan-retinal photocoagulation who were randomised to monthly ranibizumab treatment during RIDE/RISE, stratified by DRSS score (per Early Treatment Diabetic Retinopathy Study definitions7) at RIDE/RISE baseline (35/43, mild-to-moderate NPDR; 47/53, moderately severe-to-severe NPDR; 60–75, mild-risk to high-risk PDR).
Statistical analyses
Data are presented as percentages and means with 95% CIs as applicable. Between group comparisons were made using Student’s t-tests for continuous variables. Statistical significance was indicated by p<0.05.
Results
OLE patients and DRSS score improvements
Of the 500 patients who entered the OLE, 367 had DR images and non-missing data at OLE baseline and month 48. As previously reported,6 most instances of missing data at month 48 of the OLE were because of the study being terminated shortly after ranibizumab received regulatory approval in August 2012.
From OLE baseline to month 48, 72% (263/367) of patients improved or maintained their DRSS score. Among the subset of patients who had DRSS score improvement of at least one-step during RIDE/RISE (n=198), 55% (108/198) maintained or improved their DRSS score from OLE baseline to month 48, and 45% (90/198) had a worsened DRSS score.
Relationship between OLE baseline BCVA and CFT and DRSS outcomes
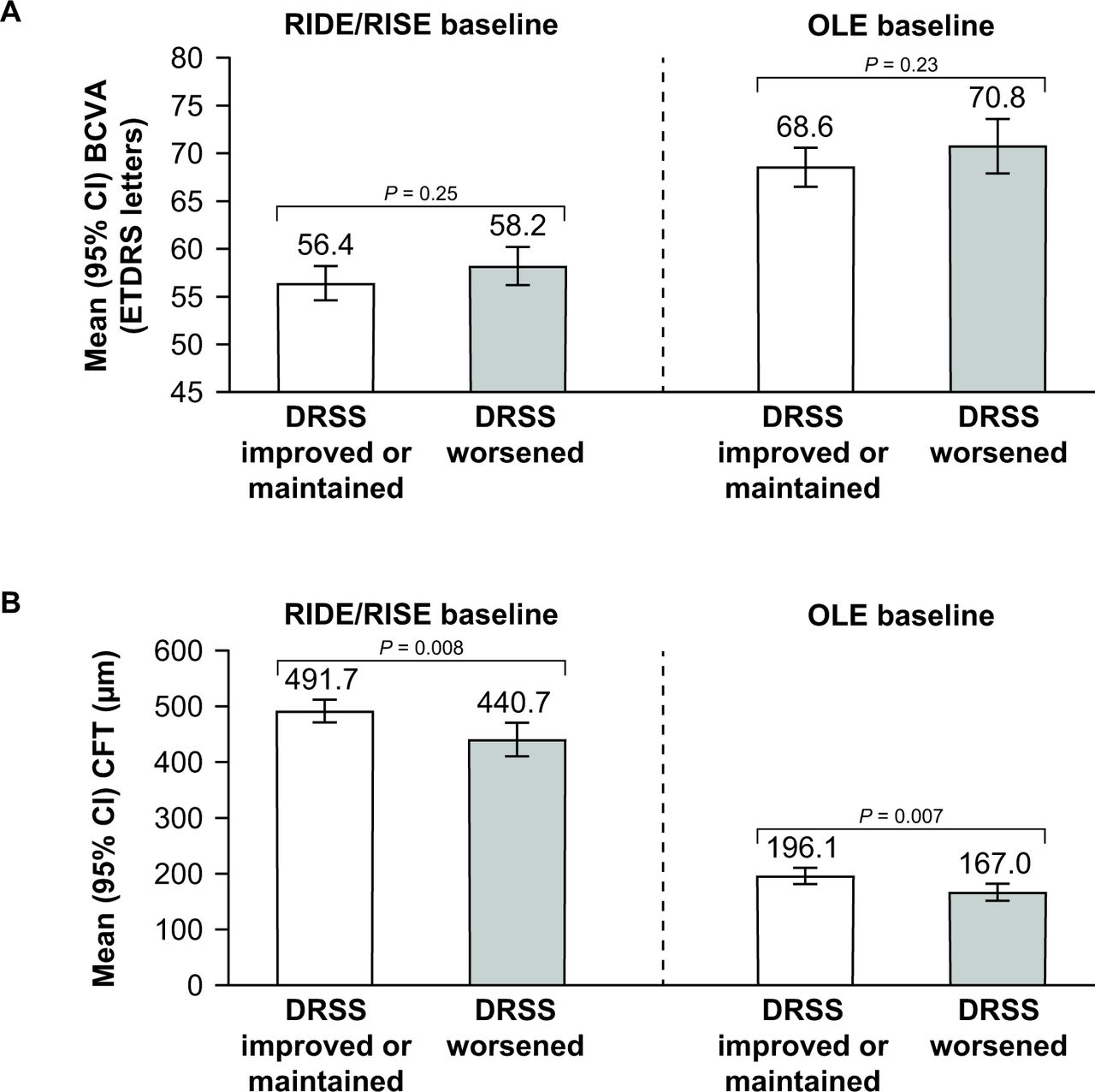
Patients who improved or maintained their DRSS score during the OLE (months 36–48) had a similar mean BCVA at RIDE/RISE baseline and OLE baseline compared with patients who had a worsened DRSS score. At OLE baseline, mean BCVA was higher (ie, better) than at RIDE/RISE baseline in both DRSS score subgroups (figure 1A).
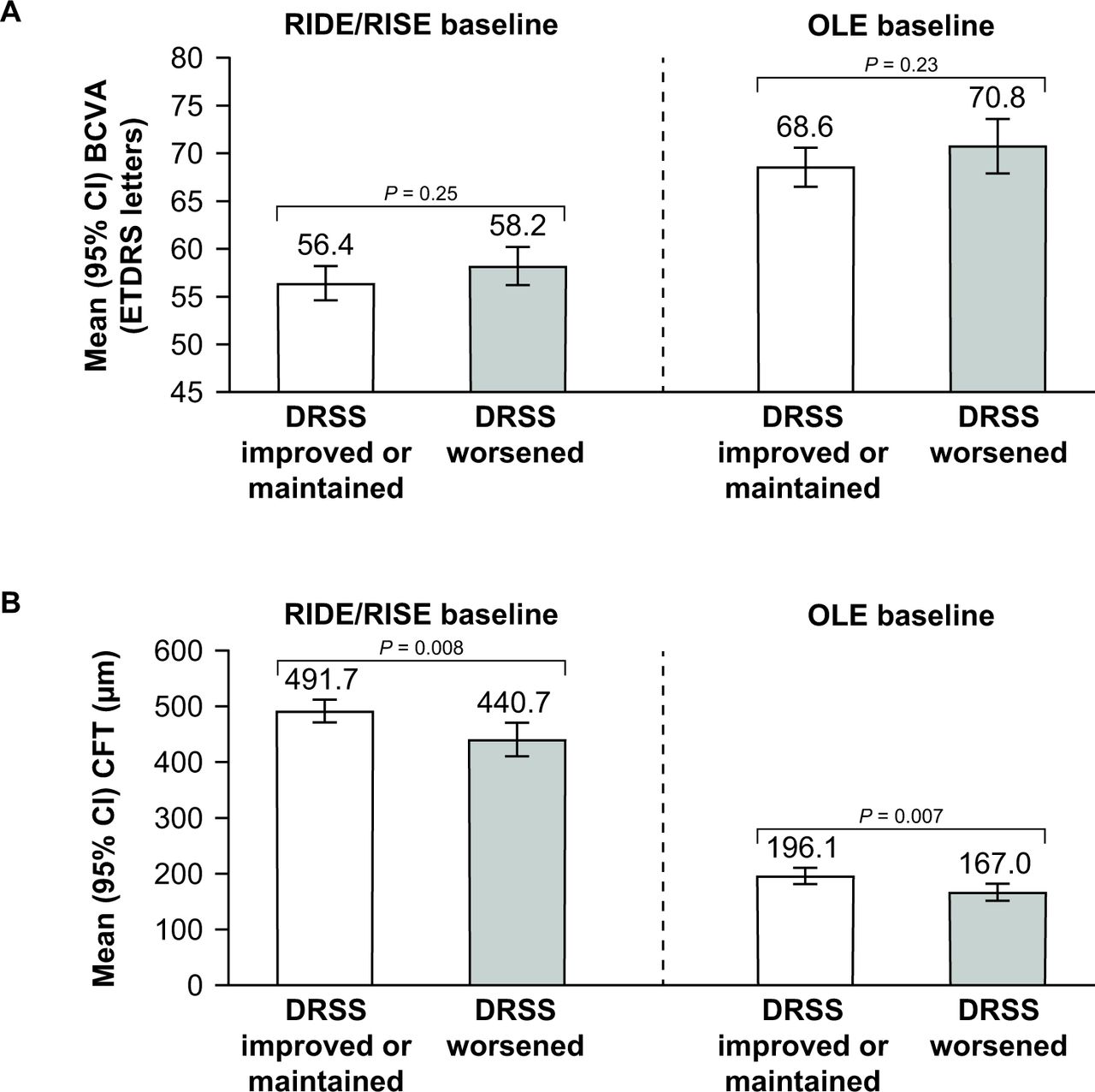
Relationship between (A) mean best-corrected visual acuity (BCVA) and (B) mean central foveal thickness (CFT) and improvement/maintenance or worsening of Diabetic Retinopathy Severity Scale (DRSS) scores at RIDE/RISE baseline and open-label extension (OLE) baseline (month 36). ETDRS, Early Treatment Diabetic Retinopathy Scale.
Patients who had an improved or maintained DRSS score during the OLE had a similar mean CFT at RIDE/RISE baseline and OLE baseline compared with patients who had a worsened DRSS score. Mean CFT was lower at OLE baseline than at RIDE/RISE baseline in both DRSS score subgroups (figure 1B). At both RIDE/RISE and OLE baseline, mean CFT was significantly lower in patients who had a worsened DRSS score compared with patients who had an improved or maintained DRSS score (both p<0.01)
DRSS outcomes by number of PRN injections during the OLE
Patients received an average of four to five ranibizumab injections between OLE baseline and month 48, regardless of baseline DRSS score (mild-to-moderate NPDR: mean, 5.0 ranibizumab injections; moderately severe-to-severe NPDR: mean, 4.5; PDR: mean, 4.2).
Patients who had an improved or maintained DRSS score during the OLE received a significantly higher (p<0.0001) number of injections (median, 4.0; mean, 4.4; 95% CI: 3.9 to 4.9) between OLE baseline and month 48 compared with patients who had a worsened DRSS score (median, 1.0; mean, 2.3; 95% CI: 1.8 to 2.9).
DRSS outcomes in native versus induced mild-to-moderate NPDR subgroups
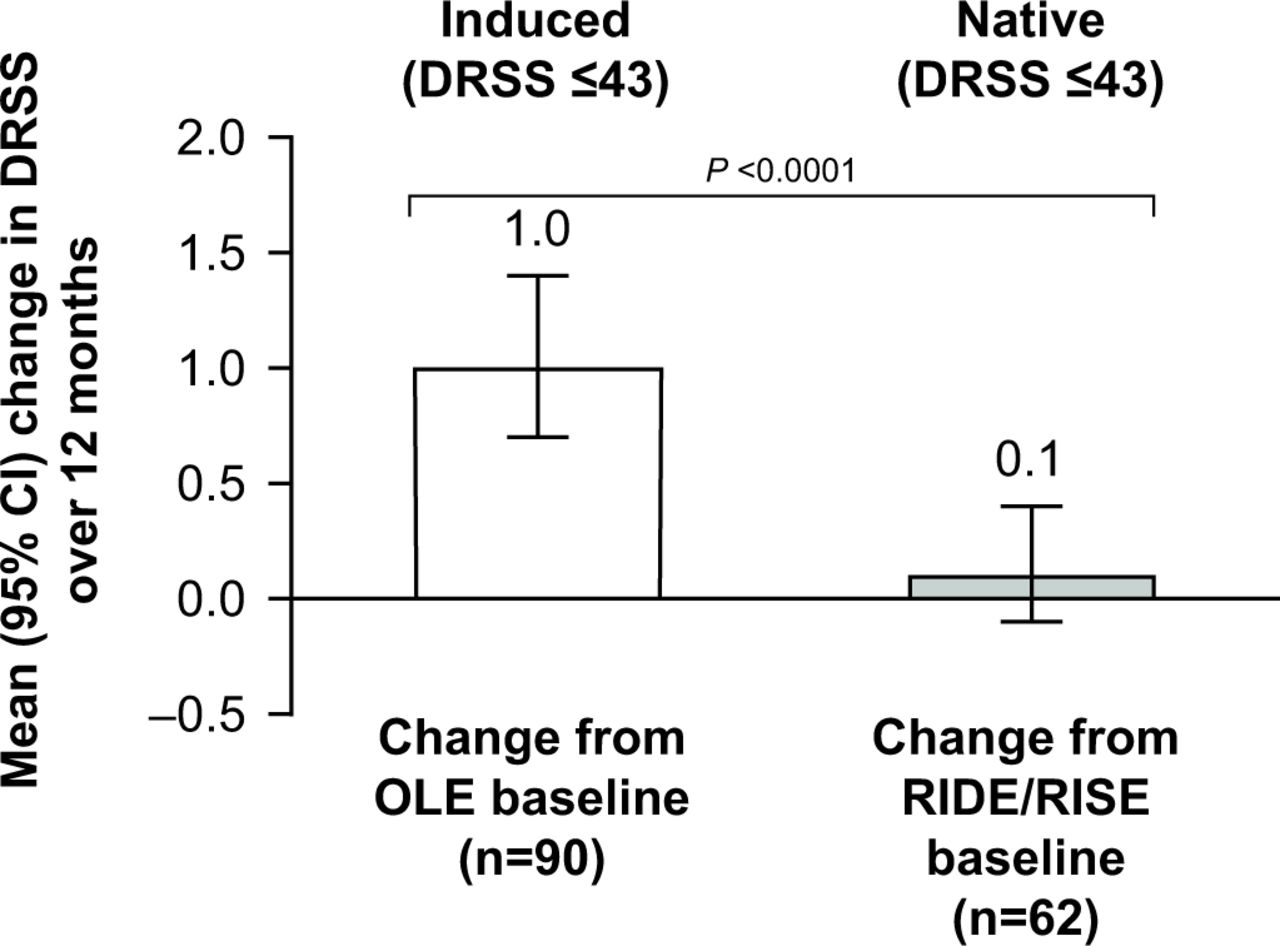
The native mild-to-moderate NPDR subgroup included 62 patients and the induced mild-to-moderate NPDR subgroup included 90 patients who had complete DRSS data at OLE baseline and month 48. Patients with induced mild-to-moderate NPDR showed significant worsening (p<0.0001) in DRSS scores from OLE baseline to month 48, with a mean (95% CI) increase of 1.0 (0.7 to 1.4) during this 12-month period, compared with a mean (95% CI) increase of 0.1 (–0.1 to 0.4) in the native mild-to-moderate NPDR group between RIDE/RISE baseline and month 12 (figure 2).

Changes in Diabetic Retinopathy Severity Scale (DRSS) scores in patients with induced and native mild-to-moderate non-proliferative diabetic retinopathy (DRSS score ≤43). Among eyes with induced DRSS score ≤43 (white column), changes were evaluated from open-label extension (OLE) baseline (month 36) to month 48. Among sham-treated eyes with native DRSS score ≤43 (grey column), changes were evaluated from RIDE/RISE baseline to month 12. A positive change in DRSS score indicates worsening of diabetic retinopathy, whereas a negative change in DRSS score indicates DR improvement.
Examination of individual patient profiles revealed that patients in the native mild-to-moderate NPDR subgroup had more stable DRSS scores than patients in the induced mild-to-moderate NPDR subgroup because more patients in the latter subgroup showed worsening in DRSS score over the 12 months of follow-up (figure 3).
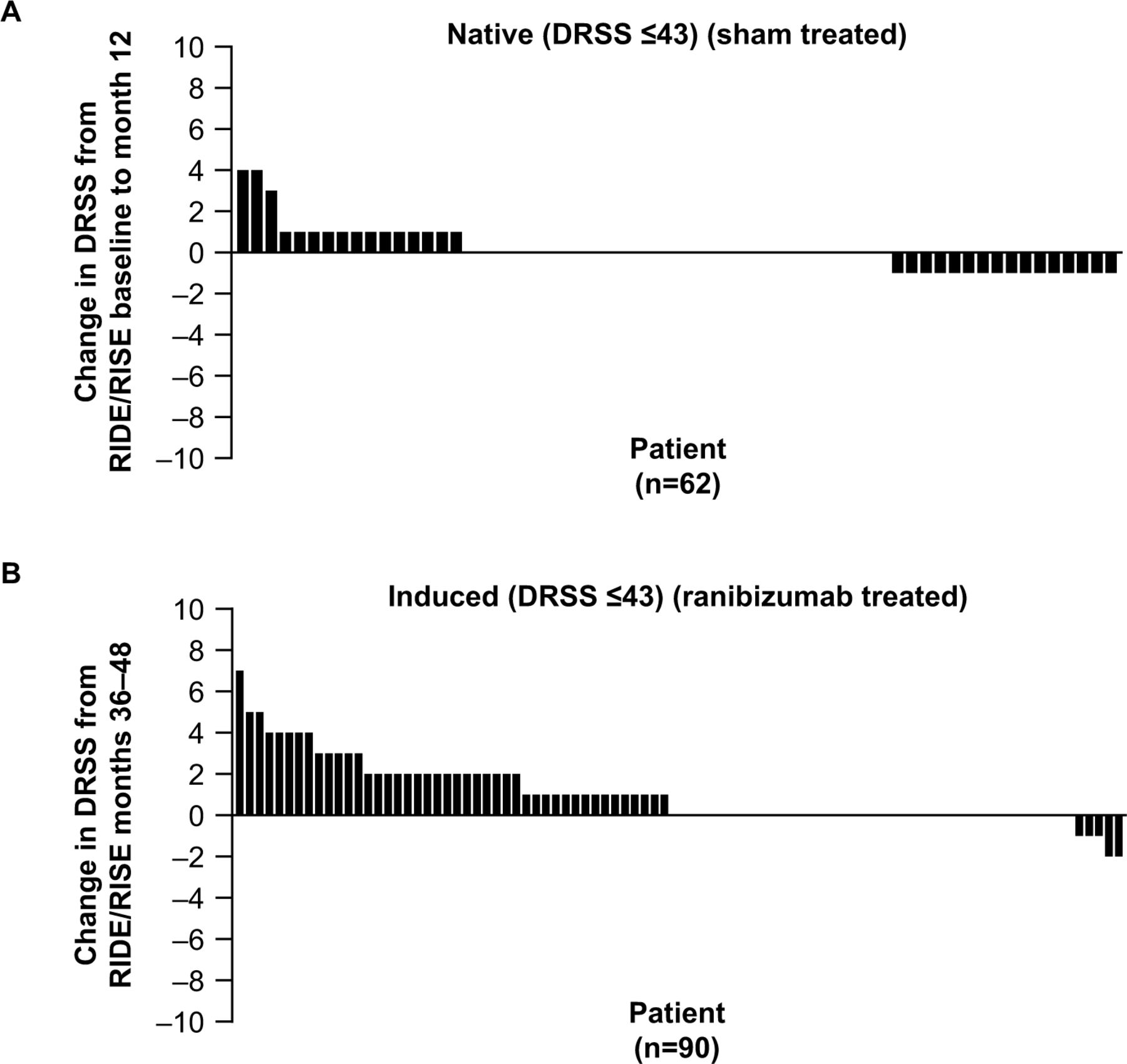
Changes in Diabetic Retinopathy Severity Scale (DRSS) scores in patients with (A) native mild-to-moderate non-proliferative diabetic retinopathy (NPDR; DRSS score ≤43) from RIDE/RISE baseline to month 12 and (B) with induced mild-to-moderate NPDR from open-label extension baseline (month 36) to month 48. Each bar represents the change in DRSS score for an individual patient. Patients are ordered from left to right by the magnitude of DRSS score change.
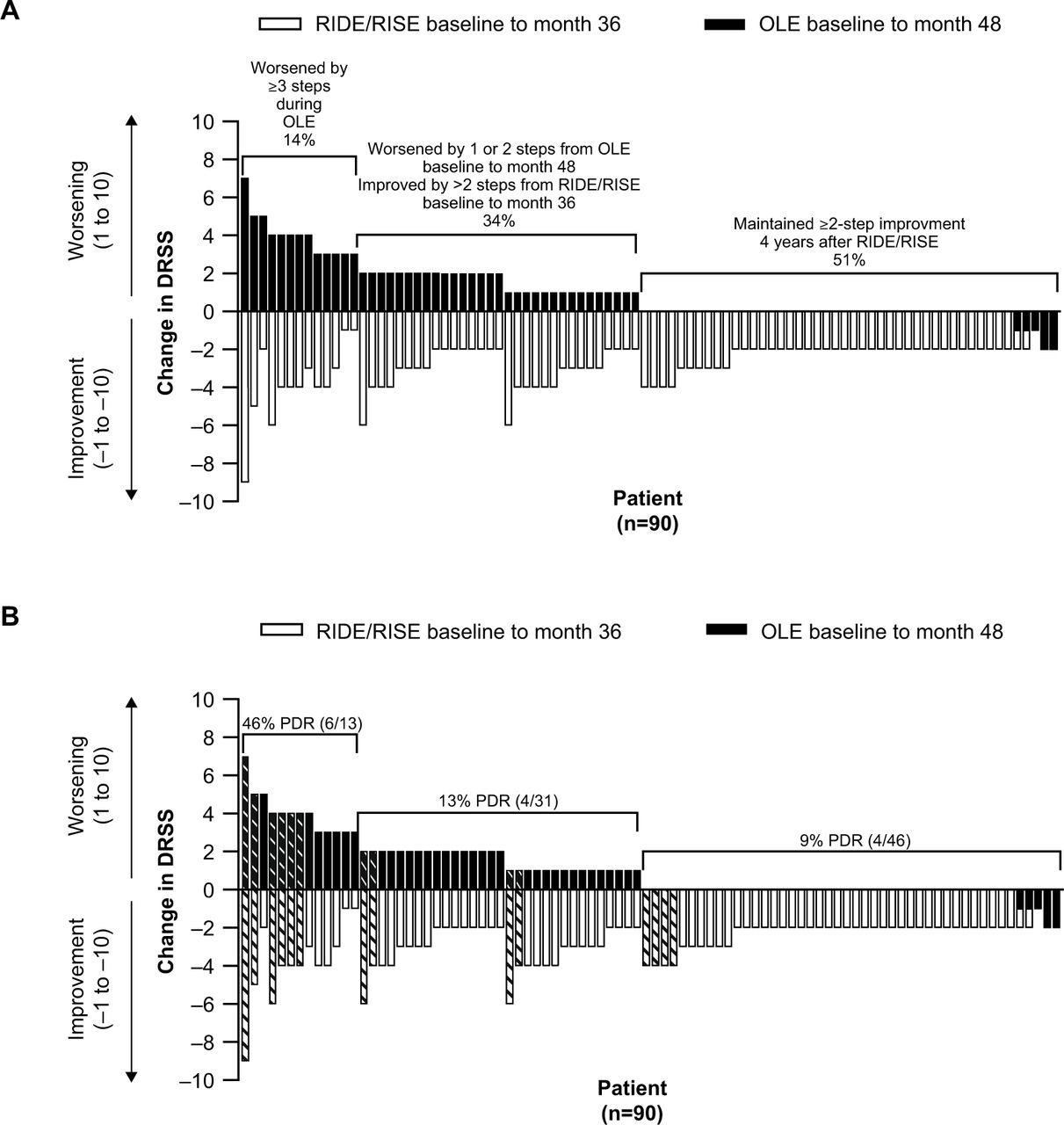
In the induced mild-to-moderate NPDR subgroup at month 48, 51% (46/90) of patients had maintained the improvement in DRSS score that was achieved during RIDE/RISE (figure 4A). A further 34% (31/90) had originally improved to DRSS score ≥43 during RIDE/RISE but worsened by one to two steps between OLE baseline and month 48, resulting in a net stable DRSS score over the entire 48-month treatment period, with improvement during RIDE/RISE and worsening during the OLE. A total of 14% (13/90) of patients had an unstable DR phenotype, in that they showed some improvement during RIDE/RISE, but then worsened by at least three steps between months 36 and 48 (figure 4A). Among patients with an unstable phenotype, 46% (6/13) had PDR at RIDE/RISE baseline (figure 4B). In contrast, only 13% (4/31) of patients with stable DR and 9% (4/46) of patients who maintained DR improvement had PDR at baseline (figure 4B).

Changes in Diabetic Retinopathy Severity Scale (DRSS) scores in patients with (A) induced mild-to-moderate non-proliferative diabetic retinopathy from RIDE/RISE baseline to month 36 and from open-label extension (OLE) baseline (month 36) to month 48, and the same plot (B) showing the distribution of patients who had been diagnosed with proliferative diabetic retinopathy (PDR) at RIDE/RISE baseline (hatched bars). Each bar represents the change in DRSS score for an individual patient. Patients are ordered from left to right by the magnitude of DRSS score change. Black bars (DRSS score change from RIDE/RISE baseline to month 36) and white bars (DRSS score change from OLE baseline to month 48) are overlaid.
Patients with PDR at RIDE/RISE baseline appeared to have the most unstable treatment responses during RIDE/RISE and the OLE compared with patients with mild-to-moderate or moderately severe-to-severe NPDR at RIDE/RISE baseline (figure 5). These patients showed the greatest improvements in DRSS score during RIDE/RISE, followed by some of the greatest deteriorations during the OLE (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in Diabetic Retinopathy Severity Scale (DRSS) scores from RIDE/RISE baseline to month 36 and from open-label extension (OLE) baseline (month 36) to month 48, stratified by DRSS score (mild-to-moderate non-proliferative diabetic retinopathy (NPDR), moderately severe-to-severe NPDR, proliferative diabetic retinopathy) at RIDE/RISE baseline. This analysis included 167 patients from the OLE who were randomised to ranibizumab treatment during RIDE/RISE and who did not have a DRSS score ≤35 at RIDE/RISE baseline and had not undergone previous pan-retinal photocoagulation. Each bar represents the change in DRSS score for an individual patient. Patients are ordered from left to right by the magnitude of DRSS score change. Black bars (DRSS score change from RIDE/RISE baseline to month 36) and white bars (DRSS score change from OLE baseline to month 48) are overlaid.
Discussion
This post hoc analysis of the RIDE/RISE OLE evaluates not only the potential predictors of changes in DRSS outcomes over time with PRN ranibizumab after a period of monthly induction therapy, but also investigates how these changes may affect observed differences in patient responses, and whether the induced regression in DRSS score behaves similar to native NPDR of the same DRSS level. These are important analyses as we seek to better understand anti-VEGF–induced regression of DR.
OLE findings from months 36 to 48 showed that the majority of patients improved or maintained their DRSS score from the initial 3-year RIDE/RISE trial period, suggesting that ranibizumab was able to provide ongoing clinical benefit on a less frequent PRN schedule. Patients with an improved or maintained DRSS score during the OLE received a median of four injections from months 36 to 48, compared with a median of only one injection in the subgroup with worsened DRSS score during the OLE. This suggests that a minimum amount of PRN treatment may be necessary to maintain any gains in DRSS score achieved during induction therapy. Importantly, BCVA or CFT, either at baseline or after induction therapy, did not affect the likelihood of maintaining DRSS score gains achieved during RIDE/RISE during the OLE, despite re-treatment criteria during the OLE being based largely on visual acuity and OCT findings.
In the current analysis, patients who had induced mild-to-moderate NPDR during the RIDE/RISE trials were significantly more prone to DRSS score worsening over 12 months of follow-up when treated intermittently with PRN ranibizumab during the OLE than patients who had native mild-to-moderate NPDR at the start of RIDE/RISE and were randomised to sham injections. This suggests that the induced improvements in DRSS score may not be equivalent to true disease regression. Indeed, the eyes with induced mild-to-moderate NPDR were more unstable than their native counterparts, and likely require ongoing aggressive VEGF suppression to prevent backsliding. Data from a retrospective analysis that used medical claims data for patients with DR who had not received anti-VEGF therapy, laser photocoagulation, intravitreal steroid treatment or retinal surgery during the baseline period support the interpretation of our data for the native mild-to-moderate NPDR subgroup. The authors of this analysis reported that the risk of DR progression and DME over 5 years was highest among patients with an initial diagnosis of moderate and severe NPDR, respectively, and that patients with mild NPDR were the least likely to progress.8 A separate post hoc analysis of data from the OLE after RIDE/RISE showed that patients who required a higher number of PRN ranibizumab injections tended to have a longer duration of DME and greater retinal thickening than those who required fewer injections; they also received more focal macular laser surgery, which was indicative of chronic disease.9
A previous post hoc analysis of RIDE/RISE demonstrated that greater treatment benefit (DRSS score improvement) over 36 months was attained with monthly ranibizumab in patients with moderate-to-severe NPDR at baseline compared with mild NPDR.10 Our post hoc analysis revealed that more severe DR at baseline was also associated with some of the greatest DR improvements during the initial RIDE/RISE trial period; however, this subgroup appeared to have less stable DR, manifested by greater worsening when monthly treatment was switched to PRN. This finding was most pronounced in patients with baseline PDR, for whom the largest fluctuations in DRSS score were observed between RIDE/RISE baseline and month 36, and OLE baseline and month 48. Of patients who exhibited some improvement in DRSS score but then worsened by three or more steps between months 36 and 48, almost half had PDR at RIDE/RISE baseline. This suggests that the significant gains acquired from monthly ranibizumab over the first 3 years of treatment might be more difficult to maintain beyond this time point with a PRN regimen if the patient has PDR to begin with, and that the patient is at high risk of worsening in the long term. Interestingly, patients in the induced mild-to-moderate NPDR subgroup received a similar number of PRN injections (four to five), irrespective of baseline DRSS score. It is possible that patients in the PDR subgroup might have benefited from an increased number of PRN injections; however, investigators did not deem this necessary at the time based on the OLE re-treatment criteria. Alternatively, our findings suggest that a period of intense treatment followed by PRN treatment may not be the best approach for patients with PDR. Other treatment approaches could be considered for these patients, including sustained VEGF suppression or scatter photocoagulation for PDR combined with ranibizumab injections for DME. The pathologic anatomical changes typical of moderate or moderate-to-severe NPDR, such as intraretinal haemorrhages or microvascular abnormalities, retinal exudates and venous beading, are more easily reversible than PDR-related neovascular changes, and the stabilisation of VEGF levels through intravitreal ranibizumab therapy may help to restore normal vascular permeability and endothelial integrity.10 Additionally, studies using OCT angiography have shown regression of neovascularisation with VEGF inhibition, and subsequent return of flow through those pathological vessels post-treatment.11 Therefore, one of the goals of anti-VEGF treatment may be to prevent progression to PDR, because this more volatile phenotype is associated with vision-threatening complications, such as vitreous haemorrhage or tractional detachment.12
Limitations of our post hoc analyses include the absence of prespecified end points or formal statistical comparisons. The original RIDE/RISE and OLE trials were not designed with inclusion and exclusion criteria to select patients and collect data that may have best addressed our hypotheses because these trials were focused on the treatment of DME, not DR. In addition, no power analyses were conducted to ensure a sufficient sample size to evaluate the data. Approximately two-thirds (500/759) of patients in RIDE/RISE continued in the OLE, but only 367 patients with evaluable DR images and non-missing data for OLE baseline and month 48 were included in the analyses. This resulted in a number of the subgroups in this analysis having relatively small numbers of patients. The main reason for missing images and data for month 48 was early discontinuation of patients after US Food and Drug Administration approval of ranibizumab for the treatment of DR. The strengths of our analyses include the detailed analysis of factors affecting the response to PRN ranibizumab treatment in the OLE and generation of hypotheses/data concerning the optimal frequency of PRN injections and the characteristics of eyes most likely to procure benefit.
In conclusion, the majority of ranibizumab-treated patients had improved or maintained DRSS scores with less-than-monthly PRN treatment. Some minimum treatment may be necessary to maintain earlier DRSS score improvement. More severe DR at baseline was associated with greater clinical benefit from monthly ranibizumab treatment during RIDE/RISE, but was also indicative of risk of DRSS score instability with intermittent dosing in the OLE. Over 12 months of follow-up, patients with induced mild-to-moderate NPDR were more likely to have worsened DRSS scores than patients with native mild-to-moderate NPDR; this may be a reflection of more severe disease at baseline and a longer time since DME diagnosis in the induced mild-to-moderate NPDR subgroup. Nonetheless, our results suggest that continuous long-term monitoring and treatment may be necessary to maintain DRSS score improvements achieved with induction therapy. Therefore, one goal of treatment of advanced NPDR may be to prevent the progression to PDR and a more unstable phenotype.
Data availability statement
Data are available upon reasonable request. For eligible studies qualified researchers may request access to individual patient level clinical data through a data request platform. At the time of writing this request platform is Vivli. https://vivli.org/ourmember/roche/. For up-to-date details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing. Anonymised records for individual patients across more than one data source external to Roche cannot, and should not, be linked due to a potential increase in risk of patient re-identification.
Ethics statements
Patient consent for publication
Ethics approval
Please note there is no approval number available. The studies (completed over 10 years ago) were carried out a multiple sites—IRB names, chairs and addresses are provided below. The RIDE/RISE study protocols (available at https://vivli.org/) were approved by institutional review boards and ethics committees at all study centres. Both studies adhered to the tenets of the Declaration of Helsinki and all participants provided written informed consent. RIDE study institutional review boards (IRBs), chairs and addresses were as follows: Copernicus Group IRB (CGIRB); Chair, Glenn C. Veit, J.D., CIP; One Triangle Drive Suite 100, P.O. Box 110605 Research Triangle Park, NC 27709. Comite de Etica Servicio de Salud Metropolitano Oriente; Chair, Andrés Stuardo Luengo, M.D. Av.; Salvador 364, Providencia, Santiago, Chile 7500922. Comité de Etica Institucional Av. Cordoba, 1830 Capital Federal, Buenos Aires Argentina C1120AAN. Comité de Etica en Investigación – Fundacion Oftalmologica Nacional; Chair, Pedro Felipe Salazar, M.D.; Calle 50 #13-50, Bogota, Colombia. Comité de Docencia e Investigación – OMI – Organizacion Médica de Investigación. Chair, Marcelo Radisic, M.D.; Calle 50 #13-50, Bogota, Colombia. University of New Mexico School of Medicine; Chair, Mark Holdsworth, Pharm.D.; Office of Research, Humans Research Protection Office MSC08 4560, BMSB Room B-71, Albuquerque, NM 87131. UCSD Human Research Protections Program; Michael Caligiuri, Ph.D.; 8905 La Jolla Village Professional Center, Suite A208 La Jolla, CA 92037. IUPUI/Clarian Institutional Review Board; Chair, Shelley D. Bizila, M.S., CIP; C/O Research Compliance, Administration, 620 Union Drive, Room 618, Indianapolis, IN 46202-5167. Comité de Etica para la Investigación de la Universidad de San Martin de Porres; Chair, Amador Vargas Guerra, M.D.; Av. Alameda del Corregidor 1531, La Molina, Lima, Peru L-12. University of Vermont Research Protections Office; Chair, Deborah Rubin, M.D.; Office of Sponsored Programs, 245 South Park Drive, Suite 900, Colchester, VT 05446. Western IRB; Chair, Michael Ognall, M.D.; 3535 Seventh Avenue, SW, P.O. Box 12029, Olympia, WA 98502-5010. Wayne State University IRB; Co-chairs, Lawrence Crane, M.D. and Michael Diamond, M.D.; 101 East Alexandrine, Detroit, MI 48201. Lahey Clinic IRB; Co-chairs, Sarkis Soukiasian, M.D. and David J. Bryan, M.D.; 41 Mall Road, Burlington, MA 01805-0002. Ochsner Clinic Foundation IRB; Chair, Joseph L. Breault, M.D., Sc.D.; 1514 Jefferson Highway CT 10, New Orleans, LA 70121. Tufts-New England Medical Center; Chair, Andreas Klein, M.D.; 750 Washington Street Tufts-NEMC #817; Boston, MA 02111. Dean Institutional Review Board; Co-chairs, R. Zorba Paster, M.D. and Daniel J. Barry, M.D., 2711 Allen Boulevard, Suite 300, Middleton, WI 53562. RISE study IRBs, chairs, and addresses were as follows: California Pacific Medical Center IRB; Chair, Gary Arsham, M.D.; 2200 Webster St, #509 San Francisco, CA 94115. Cleveland Clinic Foundation IRB; Chair, Daniel Beyer M.S., M.H.A., CIP; Cleveland Clinic Foundation Cleveland, OH 44195. Comité Institucional de Evaluación de la Facultad de Ciencias Biomédicas de la Universidad Austral; Chair, Dra. Corina Busso, President; Av. Juan Domingo Peron 1500.Derqui, Pilar.B1629AHJ. Buenos Aires, Argentina. Copernicus Group IRB (CGIRB); Co-chairs, Glenn C. Veit, J.D., CIP, Patience Stevens, M.D., M.P.H., CIP, John Falletta, M.D.; One Triangle Drive Suite 100, P.O. Box 110605 Research Triangle Park, NC 27709. Johns Hopkins Medicine IRB; Chair, Richard Moore, M.D.; Reed Hall B-130, 1620 McElderry Street, Baltimore, MD 21205. Loma Linda University Adventist Health IRB; Chair, Rhodes Rigsby, M.D., MBA; Office of Sponsored Research, 11188 Anderson Street Loma Linda, CA 92354. Massachusetts Eye and Ear Infirmary/Human Subjects Committee; Chair, Fariba Houman, Ph.D., CIP; 325 Cambridge Street, Boston, MA 02114. New York Eye & Ear Infirmary IRB; Chair, Joseph Walsh, M.D.; 310 East 14th Street, New York, NY 10003. Scott and White Memorial Hospital IRB; Chair, Matt Ridley; 2401 South 31st Street Temple, TX 76508. UMKC Adult Health Sciences IRB; Chair, Roger Sommi, Pharm.D.; 5319 Rockhill Road, Kansas City, MO 64108. University of California, Davis–IRB Administration; Chair, John Anderson, M.D.; Office of Research; 2921 Stockton Boulevard, Suite 1429, Sacramento, CA 95817. University of South Florida IRB; Chair, Berry Bercu, M.D.; Office of Research, 12901 Bruce B. Downs, Boulevard Tampa, FL 33612. University of Southern California, Health Sciences IRB; Chair, Linda Sher, M.D.; 1200 North State Street, Los Angeles, CA 90033. University of Texas IRB; Chair, Richard Rupp, M.D.; 301 University Boulevard, Galveston, TX 77555. University of Texas Southwestern Med Center at Dallas IRB; Chair, Ahamed Idris, M.D.; 5323 Harry Hines Boulevard, C1.206, Dallas, TX 75390. University of Virginia IRB for Health Science Research; Chair, Richard Stevenson, M.D.; P.O. Box 800483, Charlottesville, VA 22908. Western IRB; Chair, Theodore Schultz; 3535 Seventh Avenue, SW, P.O. Box 12029, Olympia, WA 98502-5010. Wills Eye Institute IRB; Chair, Ralph Eagle Jr, M.D.; 840 Walnut St, Suite 1020, Philadelphia, PA 19107. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank Luke Carey, PhD, CMPP, of Envision Pharma Group for third-party writing assistance, which was funded by Genentech, Inc., a member of the Roche Group.
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors RAG had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: RAG, LH, TD and IS. Acquisition, analysis or interpretation of data: RAG, LH, TD and IS. Drafting of the manuscript: RAG, LH, TD and IS. Critical revision of the manuscript for important intellectual content: RAG, LH, TD and IS. Statistical analysis: LH. Obtained funding: IS. Administrative, technical or material support: LH, TD and IS. Study supervision: IS. Guarantor: RAG.
Funding This was provided by Genentech, Inc., a member of the Roche Group, for the study and writing assistance. Genentech, Inc., participated in the study design; data management, analysis and interpretation; and preparation, review and approval of the manuscript for publication.
Competing interests RAG reports being a consultant for Allergan, Carl Zeiss Meditec, Genentech, Inc., Novartis and Regeneron; receiving lecture fees from Allergan, Carl Zeiss Meditec, Genentech Inc., and Novartis; and receiving grant support from Carl Zeiss Meditec, Genentech, Inc., Novartis and Santen. LH reports being a consultant for Alimera, Genentech, Inc., PolyPhotonix and Recens Medical. TD was an employee of Genentech at the time of the study, and is a current employee of NGM Biopharmaceuticals. IS an employee of Genentech, Inc.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.