Article Text

Abstract
Objective Idiopathic orbital inflammation (IOI) is a group of orbital inflammatory diseases of unknown etiopathogenesis. We investigated whether patients with IOI have clinical heterogeneity based on the presence (typical group) or absence (atypical group) of a unique onset that periocular inflammatory symptoms emerge suddenly but progress slowly.
Methods and analysis This retrospective cohort study included 195 patients diagnosed with IOI. We analysed the clinical data of patients, including the outcomes of corticosteroid treatment, in two subgroups stratified on the basis of the presence (130 patients) or absence (65 patients) of the unique onset.
Results Patients in the typical group were significantly younger at disease onset than those in the atypical group (median age; 52 vs 65 years, p=0.002); had more ocular adnexa-specific lesions, namely, dacryoadenitis, myositis, scleritis and optic perineuritis (78% vs 45%, p=0.00001); and had significantly fewer associations with immune-mediated inflammatory diseases (4% vs 15%, p=0.004). Among 30/119 patients (25%) who were steroid refractory in the typical group, a long period of time from symptom onset to initiation of treatment was a significant steroid-refractory risk factor (OR: 16.7), whereas, among the 18/40 patients (45%) who were steroid refractory in the atypical group, intraconal diffuse lesions were a significant steroid-refractory risk factor (OR: 8.8).
Conclusion This cohort study suggests clinical heterogeneity between the two subgroups of patients with IOI.
- orbit
- inflammation
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Idiopathic orbital inflammation (IOI) is a group of ocular adnexal inflammatory disorders of unknown etiopathogenesis.
WHAT THIS STUDY ADDS
Little information exists regarding the clinical heterogeneity of IOI. Our cohort study suggests that patients with IOI have different clinical features and outcomes following corticosteroid treatment based on the presence or absence of a unique onset that periocular inflammatory symptoms emerge suddenly but develop slowly.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE, OR POLICY
The study data and findings aid physicians in the management of patients with IOI.
Introduction
Idiopathic orbital inflammation (IOI) comprises ocular adnexal inflammatory disorders of unknown etiopathogenesis,1 2 and is the second most common disease accounting for 11%–19% of all orbital tumours.3 4 Systemic corticosteroids are considered the first-line treatment for IOI.1 2 The diagnosis of IOI has advanced through several phases. First, a panel of experts on orbital diseases presented a consensus on the criteria of IOI based on clinical indicators, imaging studies, laboratory findings and pathological indicators.1 Second, many studies have reported IgG4-related diseases in patients with IOI,2 since IgG4-related ophthalmic disease was reported.5 6
However, there are several issues concerning IOI. First, although many published articles have studied IOI as a homogeneous disease entity, clinical heterogeneity may exist in subgroups of patients. Specifically, the speed of symptom onset may be related to the underlying disease mechanism.7 Second, 38%–52% of patients are steroid refractory, but the associated risk factors have not been well determined.2 Finally, IOI also has other unique characteristics, such as migratory inflammation in orbital myositis and contralateral recurrence,2 8 and a possible relationship with immune-mediated systemic inflammatory disorders (IMIDs), such as inflammatory bowel disease and Behcet’s disease.2 9 10 However, the incidence and associations of IMIDs, such as possible IMIDs related-orbital inflammation among patients with IOI, have not been well determined.
This study aimed to investigate these three issues. In order to achieve this aim, we analysed the clinical data of patients with IOI in our patient cohort, including the outcomes of corticosteroid treatment, in two subgroups stratified on the basis of the presence or absence of a unique onset and compared them by using statistical analyses.
Patients and methods
All the patients provided informed written consent for the use of their data and the possible outcomes were explained to them.
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
This was a retrospective cohort study that included subgroup analyses. We investigated the clinical records of all the patients seen in the Ophthalmology Department of Nagoya Medical Center from 1 April 2003 to 30 June 2020, and enrolled 195 patients diagnosed with IOI. Previously published reports on IOI and orbital myositis described some of these patients.11–13 However, the clinical follow-up data were updated for this study. We recorded the patient’s age, sex, signs and symptoms, bilaterality, duration of symptoms from onset to the initial visit, past and present illnesses, and the anatomical location of the lesions. Periocular findings were recorded by pictures.
In order to compare the differences in the degree of the periocular symptoms between the two groups, we scored them based on three cardinal signs/symptoms of periocular inflammation, namely, pain, redness (including conjunctival hyperaemia), and swelling (including conjunctival chemosis). Heat was excluded from the score since the objective evaluation was challenging. In addition, mass effects, such as tumour-like lesions, were not considered swellings in our study. Based on these items, the periocular inflammation score ranged from 0 to 3 points. A score of 2 points was given to a patient presenting redness and pain. In addition, functional disturbances were scored with a maximum of 3 and a minimum of 0 points based on the presence or absence of ocular motility restrictions, ptosis, and visual acuity disturbances, including visual field defects in our study.
Based on the criteria of IOI, we defined unique onset as an acute or subacute onset in which periocular inflammatory symptoms emerge suddenly but progress slowly.1 Patients reported either the day or week of onset (eg, the third week of March). Patients also reported either these symptoms were slightly progressive before initial visits (acute onset) or they had increasing symptoms for 2 weeks after onset (subacute onset). For example, a patient who noticed eyelid swelling and then would have periocular pain 1 week after onset was classified as a subacute onset. To determine the optimal cut-off for the speed of symptom onset, our study followed the proposed criteria (cutoff >14 days).7 We also investigated the laboratory data for disease-related autoantibodies such as thyroid-associated ophthalmopathy, Sjogren syndrome and diseases with positive antineutrophil cytoplasmic antibodies, serum levels of IgG4 and other laboratory data.1 2
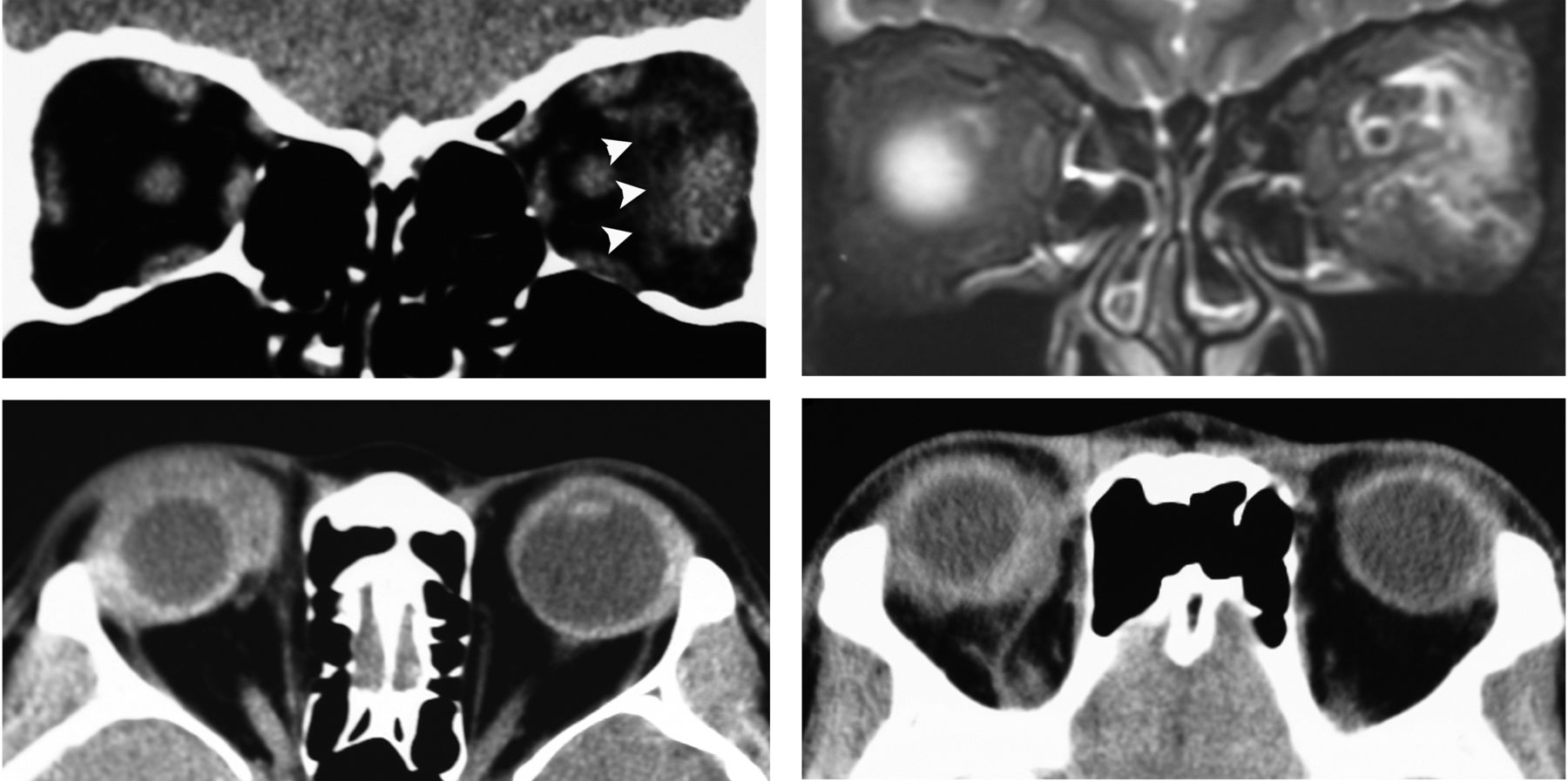
To determine the presence of ocular adnexal lesions for all patients, CT scan and MRI (figure 1) were carried out before and after treatment, and classified as diffuse IOI, dacryoadenitis, myositis, optic perineuritis, and scleritis.14 Furthermore, we subclassified the diffuse type into three subcategories: adjacent lesions of the eye (figure 1), extraconal and intraconal lesions (figure 2).
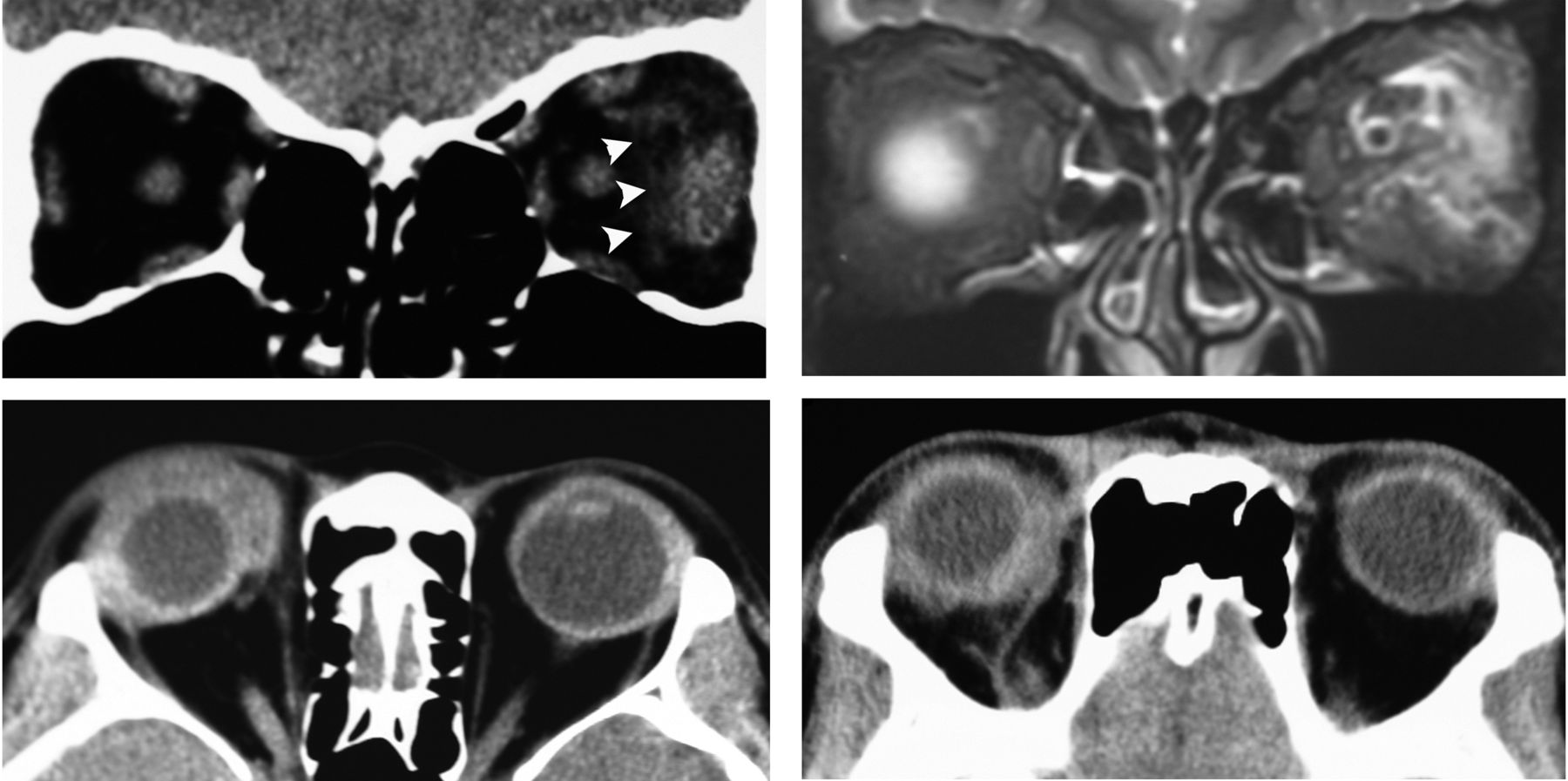
Idiopathic orbital inflammation in the typical group top: orbital myositis in the typical group. CT scan coronal view (left) shows lateral rectus muscle enlargement in the left eye and adjacent fuzzy shadows (arrowhead) and fat-suppressed T2-weighted MRI a coronal view (right) shows high signal intensity of lateral rectus muscle fascia in the left eye. Bottom: diffuse type adjacent lesion around the eye.

Idiopathic orbital inflammation in the atypical type top: CT scan (left) showing diffuse intraconal lesion in the left eye and inflammatory cells infiltrating extraocular muscle. (Right, H&E staining, original magnification ×200) bottom: Patient subsequently diagnosed with sarcoidosis. CT scan (left) showing enlargement of the right inferior oblique left and medial rectus muscles, and histopathology showing non-caseating granulomatous tissue with lymphoid cell infiltration (right, H&E staining, original magnification ×100).
To determine whether a histological confirmation for diagnosis of IOI was required, our study followed the proposed criteria.15 Thus, patients with typical myositis, scleritis and acute dacryoadenitis, which were cured within several days following corticosteroid treatment or cured within several months after observation,15 and had higher surgical risks than benefits and did not undergo open biopsy. In contrast, we performed open biopsies on 23/130 (18%) patients in the typical group and 59/65 (91%) patients in the atypical group. Histological findings were classified according to the proposed classification system, which includes classic (non-specific and polymorphous inflammatory cells infiltrations), granulomatous and sclerosing.14
We excluded the following orbital inflammatory diseases: IgG4-related ophthalmic disease,5 16–18 reactive lymphoid hyperplasia,18 19 marginal zone B-cell and other lymphomas,20 21 xanthogranuloma,22 vasculitis syndrome, thyroid-associated ophthalmopathy,23 Sjogren’s syndrome, parasitic infections,24 amyloidosis, primary and metastatic carcinomatous lesions in the other ocular adnexa,13 25 orbital cellulitis and other orbital inflammatory diseases.13 Regarding sarcoidosis, seven patients with histological granulomatous epithelioma and later confirmation of systemic sarcoidosis were excluded, and two patients with histological granulomatous inflammation and later confirmation of systemic sarcoidosis were included in the study (figure 2).
Treatments
The flow chart of the patient allocation to investigate the treatment outcomes is shown in figure 3. Systemic corticosteroid treatment was initiated only after informed consent was obtained from the patients. High dosage of corticosteroid treatment was either prednisolone at 1 mg/kg/day with a slow taper or 1000 mg intravenous methylprednisolone once per day for 3 days with prednisolone at 5–10 mg for a few weeks. Medium dosage was prednisolone at a minimum of 0.2 mg/kg/day to a maximum of 0.6 mg/kg/day with a slow taper. Patients who received prior immunosuppressive treatments due to IMIDs or other treatments, except corticosteroid treatment, were excluded from treatment outcomes. Patients in the typical group who reported less inflammatory symptoms (pain, redness and swelling) at the initial visit than those at the onset of disease and patients in the atypical group who refused treatment due to mild symptoms were observed as the natural course of IOI. Overall, we analysed 159 patients who received high-dose or middle-dose corticosteroids and 22 who were not treated.
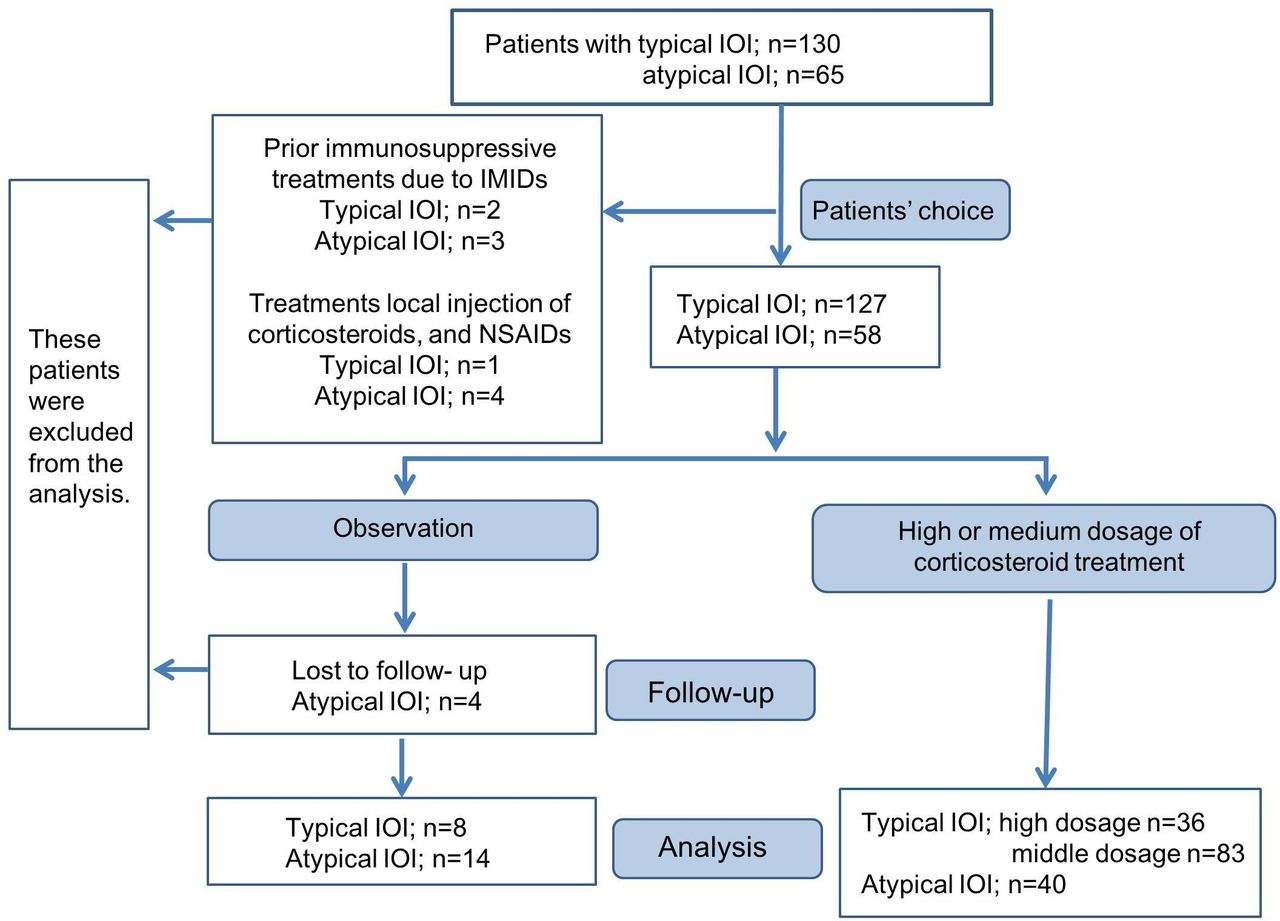
{kind=link}
{kind=link}
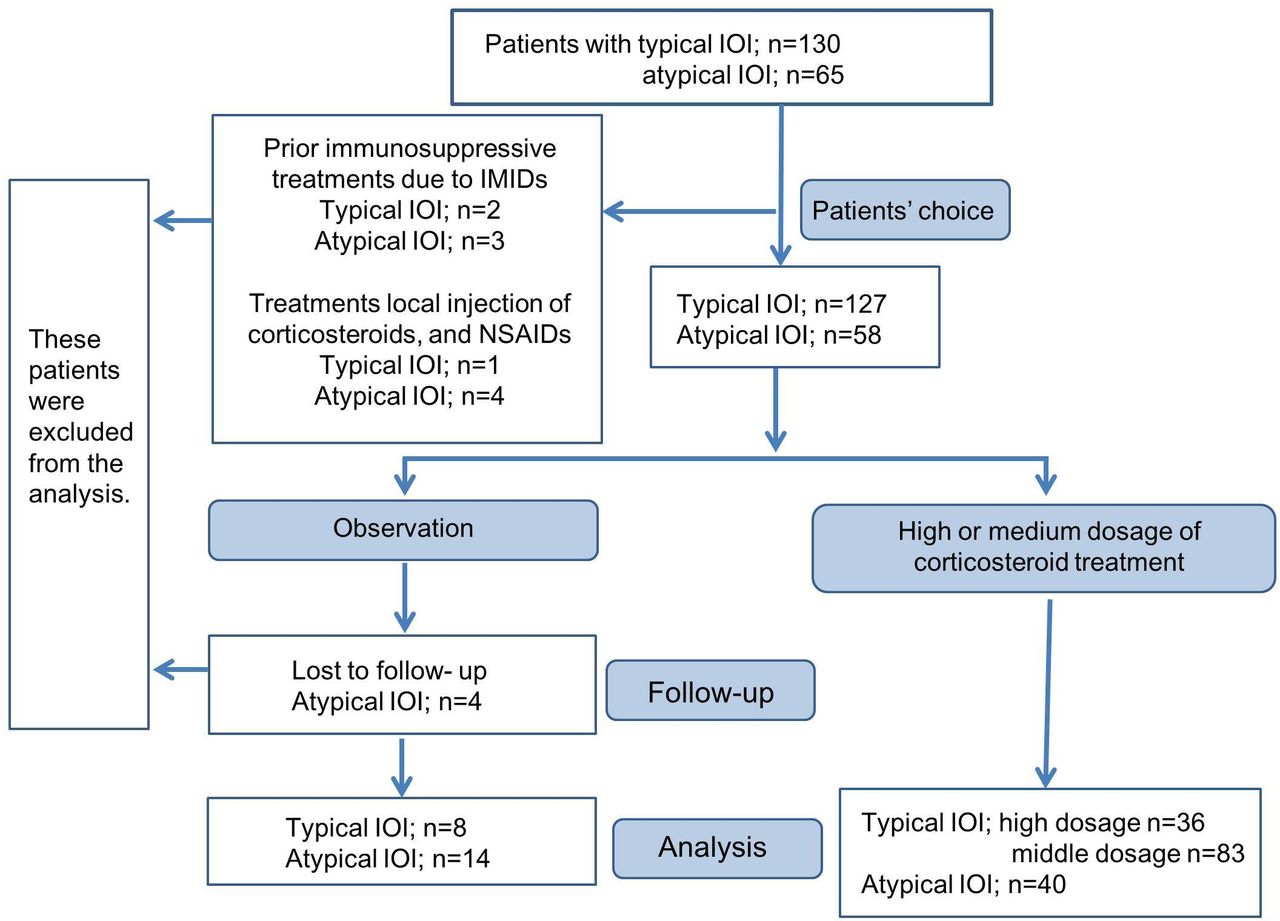{kind=link}
Flow chart of patient allocation. IOI, idiopathic orbital inflammation; IMIDs, immune-mediated inflammatory diseases; NSAIDs, non-steroidal anti-inflammatory drugs.
To assess the treatment response, we evaluated physical findings and CT scans. In addition, we followed the patients’ conditions during tapering of the corticosteroids, and determined steroid-refractory IOI, namely, incomplete treatment response, recalcitrant, and recurrent.26 When a novel ocular adnexal lesion emerged, it was considered a novel lesion. Further, we considered recurrence more than 5 years following complete remission as unpredictive recurrence and did not include it in the risk factor analysis.
Statistical analyses
To determine the differences between the two subgroups, we compared sex, a presence or absence of ocular adnexa-specific lesions, bilaterality and immune-mediated inflammatory diseases using the χ2 test. We also compared the age, number of inflammatory symptoms and functional disturbances using the Mann-Whitney U test.
To identify risk factors for steroid-refractory IOI, patients in both groups were analysed using the univariate analysis using the χ2 test and multivariate analysis using the logistic regression model.
All the statistical analyses were performed with the statistical package SPSS for Windows (V.12.0), and the statistical software EZR.27 A p<0.03 was considered statistically significant.
Results
Patient demographic data
Male patients comprised 58% (76/130) of the typical group and 45% (29/65) of the atypical group, and this difference was not significant (p=0.07). The median age of the patients was 52 (SD, 18; range 10–91 years) and 65 years (SD 18; range 11–86 years) in the typical and atypical groups, respectively. Statistical analysis showed that the typical group was significantly younger than the atypical group (p=0.002).
Considering a present or past history of IMIDs in the typical group, rheumatoid arthritis (n=1), polymyalgia rheumatica (n=1), psoriasis vulgaris (n=1), Behcet’s disease (n=1) and idiopathic thrombocytopenic purpura (n=1) were observed; meanwhile, in the atypical group, multiple sclerosis (n=1), pemphigus deciduous (n=1), idiopathic thrombocytopenic purpura (n=1), rheumatoid arthritis (n=3), eosinophilic granulomatosis with polyangiitis (n=2) and sarcoidosis (n=2, figure 2) were prevalent. The atypical group had a larger number of patients (n=10; 15%) with IMIDs than the typical group (n=5; 4%), and this difference was significant (p=0.004).
The typical group had significantly more inflammatory symptoms (maximum score 3; mean score, 2.29 vs 1.23; p<0.0001) and functional disturbances (mean 1.00 vs 0.50; p<0.0001) than the atypical group. However, neither group was significantly different between younger and older patients by inflammatory scores (p=0.9; typical group, p=0.4; atypical group). Bilaterality of the lesions between the typical and atypical groups showed no significant difference ((n=10, 8%) and (n=7, 11%), respectively; p=0.39). However, the typical group had more ocular adnexa-specific lesions (78% vs 45%; p<0.0001). Anatomical location of the lesions in the typical group were as follows: dacryoadenitis (n=20), myositis (n=57), optic perineuritis (n=10), scleritis (n=14), and diffuse type (n=29). In the atypical group the lesions were located as follows: dacryoadenitis (n=25), myositis (n=5) and diffuse type (n=35).
The predominant anatomical location of diffuse lesions varied based on the group type. In the typical group, 23 (79%) lesions were located around the eye (figure 1), 5 (18%) in the extraconal space and only one (4%) in the intraconal space. In contrast, the majority of the lesions in the atypical group were located in the extraconal space (n=15, 43%), 13 (37%) in the intraconal space (figure 2), and 7 (20%) lesions were located around the eye. The incidence of diffuse lesions located around the eye in the typical group was significantly greater than in the atypical group (p<0.0001). In contrast, the higher incidence of intraconal space diffuse lesions in the atypical group was significant (p=0.001).
We performed open biopsy for diagnosis in 23/130 (18%) patients in the typical group and 59/65 (91%) patients in the atypical group. The classification types in the typical group were as follows: classic (n=18), sclerosing (n=3) and granulomatous (n=2). Meanwhile, the histological classifications in the atypical group were as follows: classic (n=33), sclerosing (n=13) and granulomatous (n=13).
Treatment outcomes
The median time (range) from symptom onset to initiation of corticosteroid treatment for the typical was 17 days (1–256 days). A subset of patients in the typical group did not visit the hospital for long periods despite noticing the sudden onset. The atypical group sought help after 3 months (1 month to several years; three patients reported several years).
Of the 119 patients in the typical group, 89 (74%) responded well to corticosteroid treatment. Among 30 (25%) patients who were refractory to corticosteroid treatment, 16 (53%) patients (8; high dosage and 8; medium dosage) were recalcitrant and 6 (20%) patients (2; high dosage and 4; medium dosage) had an incomplete response. Eight (27%) patients had new ocular adnexal lesions (online supplemental table 1). Of the 40 patients in the atypical group, 22 (55%) responded to corticosteroid treatment. Among 18 (45%) patients who were refractory to corticosteroids, 10 (56%) patients (5; high dosage and 5; middle dosage) were recalcitrant and 8 (44%) patients (4; high dosage and 4; middle dosage) had an incomplete response. In addition, 2 (9%) patients in the atypical group showed unpredictive recurrence after 103 and 110 months (approximately 9 years). CT scan confirmed the same lesions and histology revealed inflammation classified as classical type.
Supplemental material
The results of the univariate and multivariate analyses of risk factors for steroid-refractory patterns in both groups are shown in table 1. Univariate analysis showed that in the typical group, a long period of time from disease onset to initiation of treatment (>50 days) was a significant risk factor for steroid refractory IOI (p<0.00001) and was also a risk factor by multivariate logistic regression analysis (OR, 16.7; 95% CI 5.7 to 49.3). When novel lesions were not considered steroid refractory, the OR increased (OR 17.9; 95% CI 5.9 to 54.7). Statistical analysis revealed that in the atypical group, intraconal diffuse lesion (p=0.006) was a risk factor for steroid-refractory IOI and initial corticosteroid dosage (p=0.01) was a significant negative correlation for steroid-refractory IOI; however, multivariate analysis revealed only intraconal diffuse lesion as a risk factor (OR 8.8; 95% CI 1.5 to 53). We showed the relationship between the anatomical location of lesions and histopathological findings and outcomes after corticosteroid treatment (table 2). Multivariate analysis showed that a long period of time from disease onset to initiation of treatment (>50 days) was a significant risk factor for steroid-refractory IOI (p<0.00001).
Univariate and multivariate analyses on the risk factors for steroid-refractory patients with idiopathic orbital inflammation (subgroup analyses)
Univariate and multivariate analyses on the risk factors for steroid-refractory patients with idiopathic orbital inflammation (a total of patients, N=159)
Of the 22 patients who were observed and received no treatment, all 8 patients in the typical group achieved complete regression within a maximum of 4 months (median 2 months, range 2–4 months). Meanwhile, 14 patients in the atypical group presented either an indolent clinical course (n=10) or spontaneous regression (n=4) with a median follow-up of 9 months (range 6–76 months).
Discussion
Based on the presence (typical group) or absence (atypical group) of a unique onset that periocular inflammatory symptoms emerge suddenly but develop slowly, this cohort study demonstrated clinical heterogeneity between the two subgroups considering the age of onset, degree of periocular inflammatory symptoms, the incidence of IMIDs, ocular adnexa-specific lesions, steroid-refractory factors and outcomes.
Approximately 30% of the patients in the typical group were steroid-refractory conditions that could emerge following prolonged inflammation. Thus, we suggest that patients in the typical group may have a better prognosis, when prompt corticosteroid treatment is initiated. In addition, we found that some patients in the typical group showed spontaneous recovery and novel inflammatory lesions occasionally presented in the other ocular adnexa.
Patients in the atypical group showed that the diffuse intraconal lesions were significantly steroid refractory. Published data suggest that orbital inflammatory diseases cause more fibrosis in the adipose tissues than in the lacrimal glands.28 Thus, a steroid-refractory lesion may be attributed to greater fibrosis in the adipose tissues. In contrast, this diffuse intraconal lesion may be the result of steroid-refractiveness, not the cause of it. The chronic inflammation of the intraconal lesions could adversely affect the adjacent extraocular muscles and optic nerve and involve the adjacent ocular adnexa (figure 2).
Another feature of patients in the atypical group was that patients often had fewer inflammatory symptoms. However, many patients in the atypical group did not receive treatments for longer periods before their first corticosteroid treatment. Thus, such patients may develop chronic inflammation with an increased likelihood of fibrosis and sclerosis. At this point, the inflammation is less likely to respond to steroids compared with those in acute onset inflammation treated within the first 2 weeks. However, our study did not show statistical significance of these periods. In addition, patients often showed either an indolent clinical course or spontaneous regression in a few years without any treatments. In contrast, several patients with complete regression following corticosteroid treatment developed unexpected recurrence around 9 years. A possible explanation is that these contradictory outcomes may arise from intermittently activated duration.
Further, patients in the atypical group had significantly higher associations with IMIDs in our patient cohort. A relationship between IOI and IMIDs is suggested in narrative reviews and case series studies.2 9 10 IMIDs are a group of inflammatory diseases that do not have a definitive aetiology and may be triggered by dysregulation of the normal immune response.29 Various diseases, such as Behcet’s disease, sarcoidosis, rheumatoid arthritis and inflammatory bowel disease, belong to IMIDs.30 Published data indicate that IMIDs increase the risk of inflammatory conditions in other IMIDs or chronic inflammatory diseases.31 32 In fact, patients with sarcoid-like granulomatous orbitopathy often present with systemic sarcoidosis,33 and patients with eosinophilic granulomatosis with polyangiitis and antineutrophil cytoplasmic antibody associated vasculitis occasionally present with chronic inflammatory lesions in the ocular adnexa.34 35 IMIDs have an estimated incidence of 3%–7% of the population,36 which was similar to those (4%) in the typical group. Therefore, a relationship between IMIDs and IOI in the typical group may be coincidental. However, patients in the atypical group may occasionally have IMID-related inflammatory lesions.
Interestingly, there were distinct differences between the typical and atypical groups in the diffuse type. The typical group was characterised by the surrounding eye lesions, and intraconal lesions were rare. In addition, the clinical and radiological findings of the diffuse surrounding eye lesions (figure 1) may overlap with those of scleritis. When this overlap was considered as a subtype of scleritis, the typical group had more ocular adnexa-specific lesions (95% of lesions) than initially calculated.
We did not perform a diagnostic biopsy for 82% of the patients in the typical group who met the recommendations of not requiring a biopsy,15 and did not lead to suspicions of another disease during follow-up. The indication of open biopsy has been reported in a previously published article.12 In contrast, cases of extraocular muscle enlargements without the unique onset and less periocular inflammatory symptoms revealed the presence of sarcoidosis (figure 2), IgG4-related ophthalmic disease, metastatic carcinomas, lymphoid proliferative diseases and other diseases in our hospital,5 13 17 which is consistent with the findings in the published data.37 Clinical features of the typical group in our study support the diagnostic criteria of IOI1 and may be highly specific for IOI diagnosis.
This study has many limitations. First, classification as a unique onset depends on subjective data. However, we believe that the unique onset has credibility based on research with large numbers of patients showing several statistical significances between two subgroups. Second, our study was not a prospective study regarding outcomes of corticosteroid treatment for IOI. We studied the risk factor using multivariate analyses, alternatively.38 39 Third, a small number of patients in the acute group underwent biopsy. Therefore, we could not determine the relationship between histopathological findings and corticosteroid responses. Fourth, there may be other inflammatory diseases among patients in the typical group because only a small number of patients underwent open biopsy. Fifth, published data have reported that steroid-refractory patients can respond well to infliximab.40 Radiation therapy has been considered an effective alternative treatment for IOI which is refractory to corticosteroids.2 In our study, steroid-refractory patients received additional methotrexate as an alternative treatment or low dose prednisolone for ≥6 months following repeat middle-dosage or high-dosage corticosteroids with a slow taper (data not shown). Finally, it was difficult to quantify the severity of the inflammation. Alternatively, we scored the number of inflammatory symptoms. In addition, scoring itself is subjective. However, we observed much stronger symptoms in patients in the typical group than those in the atypical group. In conclusion, considering the age of onset, degree of periocular inflammatory symptoms, the incidence of IMIDs, ocular adnexa-specific lesions, steroid-refractory factors and outcomes, our study suggests that clinical heterogeneity is based on the presence or absence of a unique onset. However, future research, such as large multicentre studies including second-line treatments, is warranted to understand IOI’s clinical features further.
Data availability statement
All data relevant to the study are included in the article or uploaded as online supplemental information.
Ethics statements
Patient consent for publication
Ethics approval
This study was performed in accordance with the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of Nagoya Medical Center, Nagoya, Japan.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Planning (TK), conduct (TK, AI), reporting (TK, AI), conception and design (TK), acquisition of data or analysis (TK, AI) and interpretation of data (TK).
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.