Article Text

Abstract
Topical fluoroquinolones (FQs) are an established treatment for suspected microbial keratitis. An increased FQ resistance in some classes of bacterial pathogens is a concern. Some recently developed FQs have an extended spectrum of activity, making them a suitable alternative for topical ophthalmic use. For example, the new generation FQs, avarofloxacin, delafloxacin, finafloxacin, lascufloxacin, nadifloxacin, levonadifloxacin, nemonoxacin and zabofloxacin have good activity against the common ophthalmic pathogens such as Staphylococcus aureus, Pseudomonas aeruginosa, Streptococcus pneumoniae and several of the Enterobacteriaceae. However, because there are no published ophthalmic break-point concentrations, the susceptibility of an isolated micro-organism to a topical FQ is extrapolated from systemic break-point data and wild type susceptibility. The purpose of this review is to compare the pharmacokinetics and pharmacodynamics of the FQs licensed for topical ophthalmic use with the same parameters for new generation FQs. We performed a literature review of the FQs approved for topical treatment and the new generation FQs licensed to treat systemic infections. We then compared the minimum inhibitory concentrations (MIC) of bacterial isolates and the published concentrations that FQs achieved in the cornea and aqueous. We also considered the potential suitability of new generation FQs for topical use based on their medicinal properties. Notably, we found significant variation in the reported corneal and aqueous FQ concentrations so that reliance on the reported mean concentration may not be appropriate, and the first quartile concentration may be more clinically relevant. The provision of the MIC for the microorganism together with the achieved lower (first) quartile concentration of a FQ in the cornea could inform management decisions such as whether to continue with the prescribed antimicrobial, increase the frequency of application, use a combination of antimicrobials or change treatment.
- Cornea
- Infection
- Pharmacology
- Treatment Medical
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Microbial keratitis (MK) is a corneal infection caused by viruses, bacteria, fungi or protozoa. It is a significant cause of preventable corneal blindness worldwide, with an estimated incidence range in high-income countries of 4.5–37.7 cases per 100 000 population-year.1 A population-based study in China estimated the prevalence of past or active infectious keratitis to be 192 (95% CI 171 to 213) per 100 000, with a prevalence of presumed viral keratitis of 110, bacterial keratitis 75 and fungal keratitis 7 per 100 000.2 3 The relative proportion of cases due to fungal infection is higher in equatorial regions.4 The risk factors for bacterial infection include contact lens wear, trauma, surgery and ocular surface disease.5 The clinical signs of infection are not a reliable indicator of the types of bacteria that are cultured,6 and there is also a delay of 24–48 hours until identification and sensitivity data become available. Therefore, it is usual to start empiric therapy with a broad-spectrum antimicrobial and then modify the treatment, if necessary, when the results of the culture and sensitivity testing against relevant antimicrobials are available. The outcome is determined by the virulence of the infecting bacteria, the susceptibility of the causative bacterium to the prescribed antimicrobial, the pharmacokinetics (PK) and pharmacodynamics (PD) of the antimicrobial and host factors such as the immune response and health of the ocular surface.7 Broad-spectrum antimicrobial cover can be achieved with a combination of two fortified and unlicensed antimicrobials, such as a beta-lactam and an aminoglycoside,8 but for the past three decades fluoroquinolones (FQs) have been used as an alternative monotherapy to provide a broad spectrum of activity against both Gram-positive and Gram-negative bacteria, Mycobacteria and anaerobes. The FQs, are a class of synthetic antimicrobials, several of which have low toxicity and are licenced for topical use.9 The advantage of monotherapy with a FQ is therefore the use of a single licensed product, with similar effect but less toxicity than a fortified aminoglycoside.10 11 Increased in vitro resistance against some FQs licensed for topical use is of concern, with the implication that monotherapy with a FQ may not be appropriate.12 13 In some regions, especially the USA, FQs have been used in combination with vancomycin to cover emerging resistance in Gram-positive isolates.12 14 In this review, we examine the properties of several new FQs that have an expanded spectrum of activity and are licensed for systemic use, some of which could be used for the topical treatment of bacterial keratitis.
Bacteria associated with MK
The bacteria isolated in cases of MK depend on the environment and regional risk factors. The proportions vary widely between reports according to the definition used to define a significant isolate. In a recent meta-analysis of 38 studies, the most common isolates worldwide were Staphylococcus spp (including Staphylococcus aureus and coagulase-negative Staphylococci) (41.4%, 95% confidence limits 36.2%–46.7%), Pseudomonas spp (17.0%, 13.9%–20.7%), Streptococcus spp (13.1%, 10.9%–15.7%), Corynebacterium spp (6.6%, 5.3%–8.3%) and Moraxella spp (4.1%, 3.1%–5.4%).15 Streptococcus pneumoniae and Nocardia spp are more frequently reported in series from South India,16–18 whereas in North Europe, Nocardia spp are rarely isolated (0.01%).19–21 The introduction of new surgical procedures may modify the profile of isolates, for example, the outbreaks of keratitis from Mycobacterium chelonae (M. chelonae), M. fortuitum and M. abscessus associated with contamination of the surgical field with non-sterile water during laser refractive surgery.22 23 Changes in the proportions of bacterial species isolated, and changes in their sensitivity to antimicrobials over time and between regions, underlines the need for continued surveillance programmes.12 24
Approach to treatment
Knowledge of the regional spectrum of isolates from bacterial keratitis can be used to guide the choice of initial antimicrobial therapy. However, there is no validated method to distinguish a pathogen from a probable contaminant and initial treatment should therefore cover the complete spectrum of common isolates.25 The choice of a monotherapy is, in part, determined by the limited number of antimicrobials licensed for topical ophthalmic use.26 The microbiological report tells the clinician whether a microorganism was identified, and whether it is likely to be susceptible or resistant to the antimicrobials relevant for topical use. Currently, the susceptibility is based on data relevant to systemic infections, which may not be applicable to topically applied antimicrobials.26 There is an association between the clinical outcome and the minimum inhibitory concentration (MIC) of the topical antimicrobial used to treat bacterial keratitis.7 27–29 Therefore, provision of the MICs of licensed and non-licensed antimicrobials for topical use could provide the clinician with more appropriate information to guide management.
New antimicrobials that are active against resistant organisms or exploit novel mechanisms of drug action are usually introduced for systemic use before they are repurposed for topical delivery.3 However, commercial development for topical use may not progress, either because the market is not large enough, the drugs are unstable or toxic to the cornea in topical form, or because they do not have sufficient advantage over existing topical antimicrobials. Inevitably, there is also a lag before most newly developed antimicrobials for are introduced for topical delivery. As a result, it is common to use unlicensed (off-label) systemic antimicrobials such as cefuroxime, ceftazidime, vancomycin, teicoplanin and meropenem to treat bacterial keratitis.30 According to the General Medical Council (UK) guidelines, prescribing unlicensed medicines may be necessary and acceptable when there are no suitable licenced alternatives, there is a supply disruption of suitably licenced drugs, when there is a serious public health risk, and if the Medicines and Healthcare products Regulatory Agency has authorised the supply of an unlicensed medicine in response.31
The relevance of antimicrobial susceptibility tests to topical therapy
The results of susceptibility testing must be interpreted in the context of a corneal infection. The MIC is the lowest concentration (mg/L) of an antimicrobial that will inhibit the visible growth of the bacterium, and the MIC90 the concentration at which≥90% growth is inhibited, within strictly controlled conditions of incubation time and temperature.3 32 33 The MIC, and other antimicrobial parameters such as dosage, PK and PD, and therapeutic success (clinical outcome), are used by regional regulatory bodies such as the European Committee on Antimicrobial Susceptibility Testing (EUCAST) or the Clinical & Laboratory Standards Institute (USA guidelines) to establish a threshold (breakpoint) concentration that determines whether the isolated bacteria is susceptible or resistant.3 The breakpoint is a chosen concentration (mg/L) of the antimicrobial that defines whether there is a high likelihood of clinical success for that agent against the isolated bacterium. If the bacterial species have an MIC below the breakpoint, they are susceptible (S), with a high probability the bacterial strain is inhibited in vivo at the concentration of the antimicrobial that is expected to be achieved at the site of the infection and, importantly, that there will be a good therapeutic response. If the bacterial species has an MIC above the breakpoint, they are resistant (R) with a high likelihood of therapeutic failure. Intermediate (I) refers to an in vitro concentration associated with clinical success with increased dosage, or relative resistance.3 Knowledge of the previous clinical response with the antimicrobial and bacterial combination is essential. For example, if an antimicrobial has a low MIC for a bacterial species but a poor clinical outcome it may be better to select an alternative antimicrobial with a better clinical response despite having a higher MIC. The reservation with this system is that the data are based on systemic administration and outcome rather than topical treatment and ophthalmic outcome. The clinical breakpoints of topically applied antimicrobials are unknown, and values based on achievable and safe serum concentrations may not be relevant.3
Although topically applied antimicrobials are delivered frequently and at a high concentration, they may still not be sufficiently biologically active in the cornea due to protein binding, changes in pH, dilution in the tear film, and continued clearance by drainage through the nasolacrimal duct.3 It is, therefore, essential to evaluate antimicrobials under conditions that model the environment of a host, including the low pH within the phagolysosome, which is particularly relevant to intracellular pathogens and infected body sites. The activity of certain classes of antimicrobials (including FQs) can be adversely affected by a reduction in the pH of the local environment.34–37
PK: ocular penetration of FQs
For a topically applied antimicrobial, the PK refers to the process by which it reaches its target site and determines the optimum dosage regimen (how much and how often) to maintain the concentration in the cornea within the therapeutic range.3 Important components include the half-life, protein binding, and the time that the concentration of the antimicrobial in the cornea remains above the MIC. Common associations with corneal ulceration, such as reflex lacrimation, inflammatory discharge, nasolacrimal duct obstruction or a keratinised ocular surface, can all affect the PK.3 Most cases of bacterial keratitis have an associated epithelial defect, but if the epithelium is intact, the drug must first traverse the lipid-rich corneal epithelium, which presents a relative barrier to hydrophilic drugs. Tight junctions between adjacent corneal epithelial cells limit paracellular transport of larger molecules. Lipophilic drugs can take a transcellular route across the corneal epithelial cells, and pores allow the passage of small (<60–100 Da) non-polar hydrophilic molecules.
The molecular weight of an antimicrobial is an important determinant of diffusion and stromal penetration. For example, the glycopeptides (vancomycin 1449.2 Da, teicoplanin 1879.7 Da) are large molecules that penetrate the intact cornea poorly, in comparison to the FQs that are much smaller molecules (eg, ciprofloxacin 331.3 Da).38 Moxifloxacin is an amphoteric lipophilic molecule that is highly soluble in aqueous with excellent corneal penetration (online supplemental file 1). Data on the PK of FQs are derived from animal models, patient undergoing penetrating keratoplasty or patients with healthy corneas undergoing cataract surgery. In these situations, with an intact epithelium, the PK is likely to be quite different to the inflamed eye with corneal ulceration.39 There are no data on the corneal and aqueous concentrations of antimicrobials from patients with bacterial keratitis. Bearing in mind these limitations, we present the available data on the corneal and aqueous concentrations of licensed FQs (online supplemental table 1). We include aqueous concentrations because this reflects the permeability of the cornea to the antimicrobial and may indicate the concentration in the posterior layers of the cornea.
Supplemental material
Pharmacodynamics
This refers to the mechanisms by which a microorganism is susceptible to a drug and the intensity of the antimicrobial effect in relation to its tissue concentration. As mentioned, the relationship between the MIC of the topically applied antimicrobial and the clinical outcome could be used to determine the ophthalmic breakpoint concentration. The clinical outcome is usually defined as time to corneal re-epithelialisation, the intensity of scar, or the spectacle-corrected visual acuity after a specified follow-up interval. Initial ulcer sizes vary, and it has been proposed that incorporating the ratio of ulcer size to the healing time could standardise outcomes.7 26–28 Several studies show a relationship between the MIC of the topically applied antimicrobial and the clinical outcome.7 27–29 Wilhelmus et al, found that infection with a bacterial strain with an MIC to ciprofloxacin greater than 1.0 mg/L was more likely to result in a lower rate of improvement and cure compared with strains with a lower MIC.28 We have shown that for Pseudomonas spp, S. aureus and Enterobacteriaceae there is a linear association between the MIC of an FQ and time to epithelial healing.3 7 In contrast, for bacteria such as Streptococcus spp and Coagulase-negative staphylococci (CoNS), there was no significant association. It is not known, however, whether this reflects the limited activity of the FQs against Streptococcus spp. However, isolates from cases of MK have also been shown to have distinctive characteristics, such as the prevalence of virulence factors that can also affect the healing time. For example, P. aeruginosa isolated from the cases of MK have a higher proportion of exotoxin U producing strains than lung isolates, with worse clinical outcomes.40
Due to limited data to establish ophthalmic breakpoints, EUCAST recommends epidemiological cut-off values as an alternative to indicate susceptibility to topical agents, determined from a comparison of the antimicrobial MIC distribution representative of the wild-type bacterial population with the MIC distribution in a population with a well characterised resistance mechanism to the antibiotic.3 Until topical ophthalmic breakpoints are established, we believe that it is preferable for the microbiology laboratory to provide the clinician with both the MIC and the bacterial susceptibility estimated from the systemic breakpoint. The clinician could then refer to the expected corneal concentration, for example, the first quartile in online supplemental table 1, and decide if the MIC is above or below that concentration. Providing the MIC to the clinician has distinct advantages. For example, if the MIC is close to the achievable concentration of the antimicrobial in the cornea the options would be to increase the dosage (eg, frequency of application), change the route of application (eg, intrastromal injection), change the antimicrobial or add a second antimicrobial to achieve an additive or synergistic response. In contrast, if an antimicrobial has a low MIC for a bacterial isolate but has a poor clinical response, it may be better to select an alternative antimicrobial with a better clinical outcome even though it has a higher MIC.3 The MICs of FQs to the bacteria commonly isolated from bacterial keratitis are presented in table 1.
MICs of antimicrobials in bacterial keratitis
Fluoroquinolones
In the early 1960s, Lesher et al, reported the potential of 7-chloro-quinoline, a precursor of the antimalarial chloroquine, as an antimicrobial,41 with nalidixic acid introduced as the first quinolone antimicrobial.42 Since the 1990s, FQs have become a popular bactericidal treatment for bacterial keratitis figure 1 due to their availability as a licensed product, low toxicity and broad spectrum of activity.43

The bicyclic core structure of fluoroquinolones. X and Y are carbon or nitrogen atoms. A carbon atom at Y defines the quinolones. Fluoroquinolones have a fluorine (F) atom at C6. Different substitutions at positions R1, R5, R7 and R8 can improve the activity of the drug. Adapted from Pham et al and Rusu et al 42 44
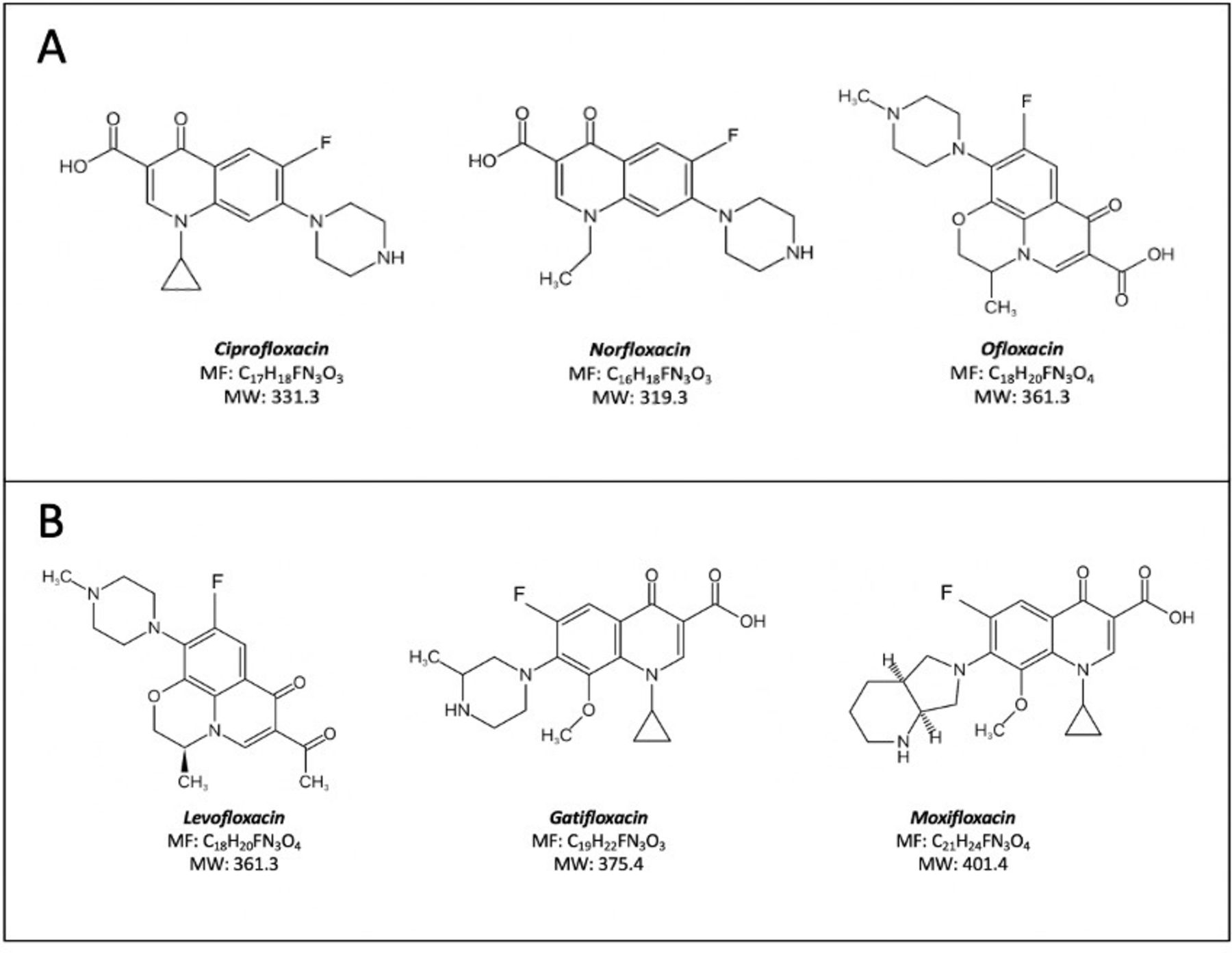
The quinolones have been grouped into four generations according to their structure and spectrum of activity. The first-generation quinolones were pipemidic acid, piromidic acid, oxolinic acid and cinoxacin. They are not used in ophthalmology. The second-generation quinolones started with flumequine with a fluorine atom in the C6 position of the nucleus, a characteristic of FQs, which significantly extended their spectrum of activity to include Gram-positive as well as Gram-negative bacteria.42 44 Other second generation FQs include ciprofloxacin, norfloxacin and ofloxacin (figure 2A).

Structures of second, third and fourth generation fluoroquinolones. Molecular formula (MF), Molecular weight (MW in Daltons). A. Second generation fluoroquinolones. B. Third (levofloxacin and gatifloxacin) and fourth (moxifloxacin) generation fluoroquinolones
Third generation FQs, such as levofloxacin and gatifloxacin, have an increased activity against Gram-positive bacteria (Streptococcus spp), better tissue penetration and a longer half-life, which allows increased dosing intervals.44 In the fourth generation FQs the addition of a nitrogen or a methoxy group at the R8 position improved anaerobic cover (see moxifloxacin figure 2B).42 44 Not all newest (fifth generation) quinolones have a fluorine atom, which is not essential for the antimicrobial effect, so they are technically not FQs. They are not licensed for ophthalmic use but include levonadifloxacin, nadifloxacin, nemonoxacin, finafloxacin, zabofloxacin, avarofloxacin (acorafloxacin), lascufloxacin and delafloxacin (see figure 3).45 46

Structures of newest fluoroquinolones. Adapted from Rusu et al. 42
(A) The following FQs are currently licensed for topical ophthalmic use.
Ciprofloxacin, ofloxacin, levofloxacin and norfloxacin are second or third generation FQs used as empirical treatment for bacterial keratitis and conjunctivitis. In randomised controlled trials, ciprofloxacin and ofloxacin were as effective and less toxic than fortified aminoglycoside (1.5% gentamicin or 1.3% tobramycin) and a beta-lactam (5% cefuroxime or 5% cefazoline).11 ,45 Levofloxacin, a third-generation FQ that is the L-isomer of ofloxacin (see figure 2), has good activity against gram-positive bacteria such as Streptococcus pneumoniae but less activity against gram-negative organisms such as P. aeruginosa. Due to the improved activity of levofloxacin and the newer FQs against Gram-positive bacteria, the topical use of ciprofloxacin and ofloxacin has reduced.13
Moxifloxacin is a fourth generation FQ with an extended spectrum of activity against Gram-positive organisms such as S. pneumoniae. Because it binds to both bacterial gyrase and type IV isomerase, it is less likely to select resistant strains.46 47 It is highly lipophilic and very soluble in the aqueous humour at a physiological pH, so it rapidly establishes a concentration gradient across the cornea.48 49 Experimentally, moxifloxacin achieves a significantly higher aqueous concentration when applied topically in comparison to the oral route.48 The penetration of moxifloxacin significantly exceeds ciprofloxacin when assessed as the concentration in the aqueous humour of patients undergoing cataract surgery.50 It should be noted, however, that in this study, the standard deviations were high compared with the mean concentration, and this finding should be treated with caution. For example, the first quartile of the corneal concentration may be below the MIC of the bacterial isolate (see online supplemental table 1). There is quite marked variability in the reported corneal and aqueous concentrations of moxifloxacin, and it has relatively high (50%) protein binding, which may reduce bioavailability (table 2, online supplemental file 1). Two small randomised clinical trials have compared 0.5% moxifloxacin or 0.3% gatifloxacin with combination therapy of 1.3% tobramycin and 5% cefazolin, or ofloxacin with tobramycin and cefazolin.51 ,52 They showed no statistically significant difference in healing time between the groups. An increase in moxifloxacin resistance in Streptococcus spp, methicillin-resistant S. aureus (MRSA) and Pseudomonas spp has been reported in some countries.12 53
Pharmacokinetic features of fluoroquinolones
Besifloxacin 0.6% is a fourth generation FQ licensed in some countries to treat bacterial conjunctivitis. It was the first FQ introduced exclusively for ophthalmic use.42 54 Its structure includes a chlorine substituent at the C8 position and an amino-azepinyl group at the C7 position,54 which enables dual inhibition of DNA gyrase and topoisomerase IV.55 Besifloxacin has superior in vitro activity against S. pneumoniae gyrase and topoisomerase IV compared with ciprofloxacin and moxifloxacin,55 which broadens the spectrum of activity while reducing the risk of antimicrobial resistance (AMR).56 57 Besifloxacin also has anti-inflammatory properties and inhibits proinflammatory cytokines in vitro, although the clinical relevance of this is unknown.58 In a post hoc in vitro analysis of the MIC90 of isolates from three clinical trials of the treatment of bacterial conjunctivitis, besifloxacin was reported to show lower MICs than moxifloxacin, gatifloxacin, levofloxacin and ciprofloxacin against Gram-positive isolates, most notably ciprofloxacin-resistant Staphylococcus isolates, and similar potency to moxifloxacin against Gram-negative bacteria.59 To date, however, there is no randomised controlled trial evidence to support its use in bacterial keratitis.
(B) The following FQs are potentially suitable for topical ophthalmic use.
Delafloxacin
Delafloxacin is a novel FQ modified to broaden its spectrum of activity.60 It behaves as a weak acid due to its 3-hydroxyazetidinyl moiety at position 7 of the aromatic ring (see figure 3).42 61 62 This moiety distinguishes delafloxacin from other FQs as it can accept protons in acidic conditions and rapidly traverse cellular membranes at an acidic pH. On entering the cell, it loses its proton and returns to the anionic state.61 This property increases intracellular potency in acidic conditions when compared with ciprofloxacin and moxifloxacin, as may occur in inflammation and abscesses.63 Experimentally, delafloxacin has better uptake in bacteria and eukaryotic cells, associated with an increased activity against S. aureus and MRSA.61 64 Similar to the fourth generation FQs, delafloxacin is active against both bacterial gyrase and topoisomerase IV, reducing the risk of AMR.65 In a recent in vitro study comparing delafloxacin to levofloxacin, moxifloxacin and vancomycin against bacterial isolates from cases of endophthalmitis, delafloxacin showed significantly lower rates of resistance to S. epidermidis and S. aureus, including methicillin-resistant isolates, with low MICs.66 Direct comparison with moxifloxacin across all the Staphylococcus isolates from this study demonstrated significantly increased susceptibility to delafloxacin. Delafloxacin has low (16%) protein binding, suggesting that its activity may not be significantly reduced in the inflamed cornea. To the best of our knowledge, there are no published data on the clinical use of topical delafloxacin for bacterial keratitis, but it is a good candidate for evaluation.

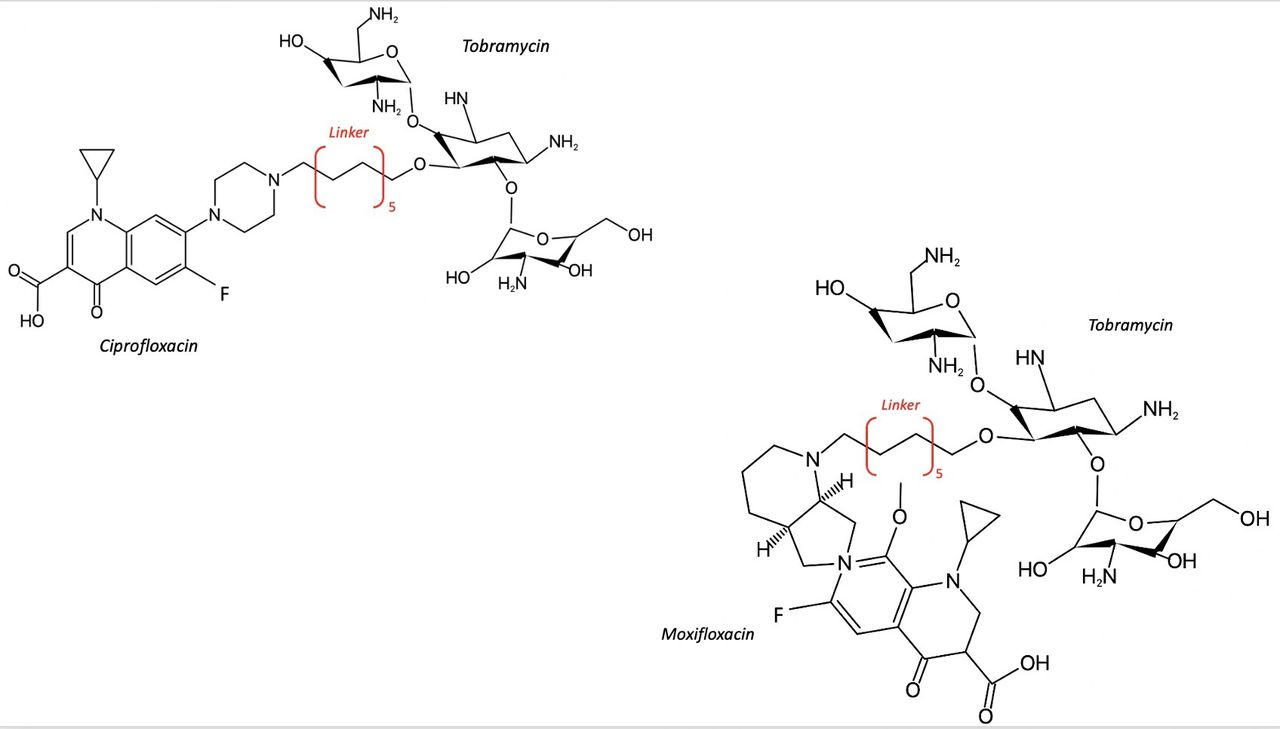
Structures of tobramycin-ciprofloxacin and tobramycin-moxifloxacin hybrids. Re-drawn from Domalaon et al. 81
Nadifloxacin and levonadifloxacin
Levonadifloxacin is a novel antimicrobial that belongs to the benzoquinoline subclass of quinolones. It has a potent bactericidal action and has improved activity in acidic environments.67 Levonadifloxacin is active against the clinically significant Gram-positive, Gram-negative and anaerobic organisms isolated from respiratory infections, such as methicillin-resistant and quinolone-resistant S. aureus, S. pneumoniae, S. pyogenes, H. influenzae and Moraxella catarrhalis and atypical pathogens.68–73 There is also activity against quinolone-susceptible Gram-negative bacteria including Escherichia coli, Klebsiella pneumoniae, Pseudomonas spp and Acinetobacter spp. Furthermore, after systemic administration, levonadifloxacin has inhibitory action against proinflammatory cytokines.74 Due to their antibacterial and anti-inflammatory properties, they are used topically for the treatment of acne vulgaris and may have a role in the role of ocular rosacea and related lid disease.75 There are no data regarding its use as a topical ophthalmic medication, but it is a promising candidate.
Ozenoxacin
Ozenoxacin is a non-fluorinated quinolone with dual activity against bacterial gyrase and topoisomerase IV and currently used in the treatment of impetigo.76 It has excellent bactericidal activity against Methicillin-sensitive S. aureus, MRSA, MRSE, S. pyogenes, P. acnes and ofloxacin-resistant strains of S. aureus and S. epidermidis .77 The MIC90 against P. aeruginosa, however, is almost double that of ofloxacin and levofloxacin, which may limit its utility for the treatment of suspected bacterial keratitis.77
Nemonoxacin
Nemonoxacin is a novel, C-8-methoxy non-fluorinated quinolone with both oral and IV formulations. It has a broad spectrum of activity with low toxicity.63 It has a safety profile similar to levofloxacin when treating community-acquired pneumonia (CAP). Although it has low water solubility, its low protein binding (16%) may make it an option for the topical treatment of bacterial keratitis.78
Zabofloxacin
Formulations of zabofloxacin are available as zabofloxacin hydrochloride (DW-224a) and aspartate (DW-224aa), both of which provide bactericidal action against Gram-positive and Gram-negative bacteria, including the Enterobacteriaceae. Furthermore, these FQ’s have been shown to be effective against quinolone resistant strains, although there are no data on topical use for bacterial keratitis.63
Avarofloxacin
Avarofloxacin (JNJ-Q2) is an aminoethylidenylpiperidine FQ that is an effective antistaphylococcal agent.79 Its zwitterion structure imparts an antimicrobial effect against numerous Gram-positive bacteria with an MIC90 value of 0.12 mg/L, which is more potent than other FQs (table 3). It is also active against Strep. pneumoniae, MRSA, Enterococcus spp, E. coli, Klebsiella spp, H. influenzae and P. aeruginosa (table 3). The absorption and permeability properties of avarofloxacin may be similar to the currently approved FQ’s due to its low molecular weight, solubility and lipophilicity. There are no data on topical use for bacterial keratitis.
MICs of novel fluoroquinolones to systemic (non-keratitis) isolates
Lascufloxacin
Lascufloxacin is approved in Japan to treat respiratory infections including, but not exclusive to, community acquired pneumonia and ear, nose and throat infections. Lascufloxacin is a hydrochloride salt (oral formulation, Lasvic 75 mg tablets). Lascufloxacin inhibits DNA synthesis by binding to DNA gyrase and topoisomerase IV.42 It has higher tissue penetration than levofloxacin or moxifloxacin. It is effective against Gram-positive bacteria, including usually resistant species such as S. pneumoniae first step mutations. There are no data on topical use of lascufloxacin, but it is insoluble in water or dimethyl sulfoxide, so an aqueous formation may not be possible.
Finafloxacin
Finafloxacin is a fluorinated quinolone derivative with an 8-cyano substituent and pyrrolo-oxazinyl moiety. It has increased efficacy under acidic conditions such as skin and soft tissue, vagina and urinary tract. Although the pH of the corneal surface reduces when the eye is closed, the pH in cases of bacterial keratitis is unclear. In vitro, finafloxacin has activity under acidic and neutral mediums against bacteria, including S. aureus and Acinetobacter baumannii. The activity at a range of pH values may, therefore, be an advantage compared with antimicrobials such as ciprofloxacin that may have reduced activity in an acidic environment.80 There are no research data on its protein binding properties and its potential mode of action in the cornea is extrapolated from in vitro studies. At present finafloxacin is licensed as a treatment for otitis externa. Garenoxacin was patented as an eye-drop but, as far as we are aware, it was not developed for clinical use.
MICs of FQs
Where data are available, we show the MIC for the FQs licensed or unlicensed for treatment of MK in tables 1 and 3. The MIC90 of keratitis isolates are generally less than the first quartile concentration (online supplemental table 1), but the variation depends on the species. In general, the maximum concentration achieved in the stroma should exceed the MIC by a factor of 4. For example, if the MIC of S. aureus to moxifloxacin is 2 mg/L, then a corneal concentration of moxifloxacin should be ≥8 mg/L. It is not clear from the literature whether bacteria can invade the anterior chamber to cause endophthalmitis in the absence of a perforation, However, it is reasonable to aim at an aqueous concentration greater than the MIC when treating bacterial keratitis.
Factors affecting bioavailability of FQs
Although bioavailability, urinary fraction and maximum serum concentration are important for systemically administered antimicrobials, protein binding and other local environment factors such, as pH or release of ions, which might interfere with an antimicrobial’s activity, are of more importance for topically administered antimicrobials. This is evident in the lower bioavailability (activity) of an FQ such as ciprofloxacin in the cornea compared with its chemical concentration, which is the parameter that is usually measured.33 Compared with ciprofloxacin and levofloxacin, delafloxacin and nemonoxacin are much less affected by protein binding (table 2). Therefore, they are expected to have greater bioavailability in the cornea in MK.
FQs that are not licensed for topical ocular use must be compounded into a suitable preparation. There are a variety of topical ophthalmic formulations available, including aqueous or oily solutions, suspensions, emulsions, gels and colloidal systems. Reformulation can be based on a licensed systemic FQ as the starting raw material but requires factoring in the chemistry of the active ingredient so that the required concentration to deliver the intended antimicrobial activity is sustained. This may require adding excipients into the preparation to adjust pH, tonicity and viscosity. pH adjustment may improve the stability and solubility of the active agent and reduced crystallisation. Once formulated, eye-drops may be packaged in either ‘single use’ or multidose containers to maintain drug concentration and stability. This will increase the probability that the antimicrobial will meet the required antimicrobial effectiveness test. An antimicrobial preservative is often added to multidose containers, although some self-preserved medicines such as moxifloxacin may pass these tests without preservative addition. Reformulation of systemic FQs for topical application must comply with good manufacturing standards within a suitably approved compounding unit.
FQ combination therapy
The success of treatment of bacterial keratitis depends on the MIC of the antimicrobial against the infecting organism, and there are strategies to produce lower MICs through antimicrobial combinations. The results can be synergistic, additive, indifferent or antagonistic (table 4). Synergistic combination therapy, in which two antibiotics are used simultaneously with enhanced effect, has the advantage of lower MICs, broader antimicrobial cover and reduced AMR selection pressures. For example, the use of gentamicin in combination with penicillin in the treatment of Enterococcus is synergistic; the penicillin-mediated disruption to the bacterial cell wall promotes the uptake of gentamicin, resulting in enhanced bactericidal effects. When there is antagonism, two antimicrobials used simultaneously inhibit one another, for example, the combination of chloramphenicol and penicillin is antagonistic in the treatment of meningococcal meningitis resulting in significantly higher mortality. Additive indicates that the combination results in a lower MIC than each of antimicrobials. The measure used to classify these effects is the fractional inhibitory concentration (FIC) index, where the FIC is determined for each antimicrobial by dividing the MIC for each agent when used in combination by the MIC of each agent when used alone. For example, synergy is defined when the sum of FIC for each antimicrobial is ≤0.5 while antagonism is defined when the sum of the FICs>4. In current ophthalmic practice, combination therapy for MK is frequently used to provide broader antimicrobial cover as opposed to synergy. In contrast, sequential treatment with chloramphenicol often follows a course of treatment of bacterial keratitis.
Synergy and antagonism with fluoroquinolone combination therapy
Of the topical antimicrobial combinations tested in vitro, meropenem and either ciprofloxacin or moxifloxacin have a synergistic response. For S. aureus and P. aeruginosa isolates, there was synergy in 10% of isolates (table 4). An additive effect was demonstrated in 70% of the remaining S. aureus and 80% of the P. aeruginosa isolates (table 4). Although gentamicin and moxifloxacin also produce a synergistic effect against 20% of P. aeruginosa isolates from MK, this may not occur in vivo as gentamicin has poor cellular penetration. Finally, although it is unknown whether these results translate into improved clinical outcomes, a lower MIC has been associated with a faster healing response. There are numerous other combinations worthy of investigation.
Antimicrobial hybrids formed by covalent bonding of active pharmacophore fragments into cleavable (prodrug) or non-cleavable structures were proposed to reduce AMR and most hybrids contain a FQ pharmacophore. The combination of pharmacophore fragments into one heteromeric compound results in a single PK profile and may impart synergistic effects or even produce a new mechanism of action. Antimicrobials struggle to penetrate the outer membrane of P. aeruginosa. As such tobramycin hybrids were developed based on the increased uptake of an aminoglycoside to deliver a second antimicrobial into the periplasm of a Gram-negative microorganism such as P. aeruginosa. Examples are tobramycin linked to ciprofloxacin or moxifloxacin for the treatment or P. aeruginosa infections (figure 4). Further investigation into the application of hybrids for use as topical therapy is warranted.81
Mechanism of action
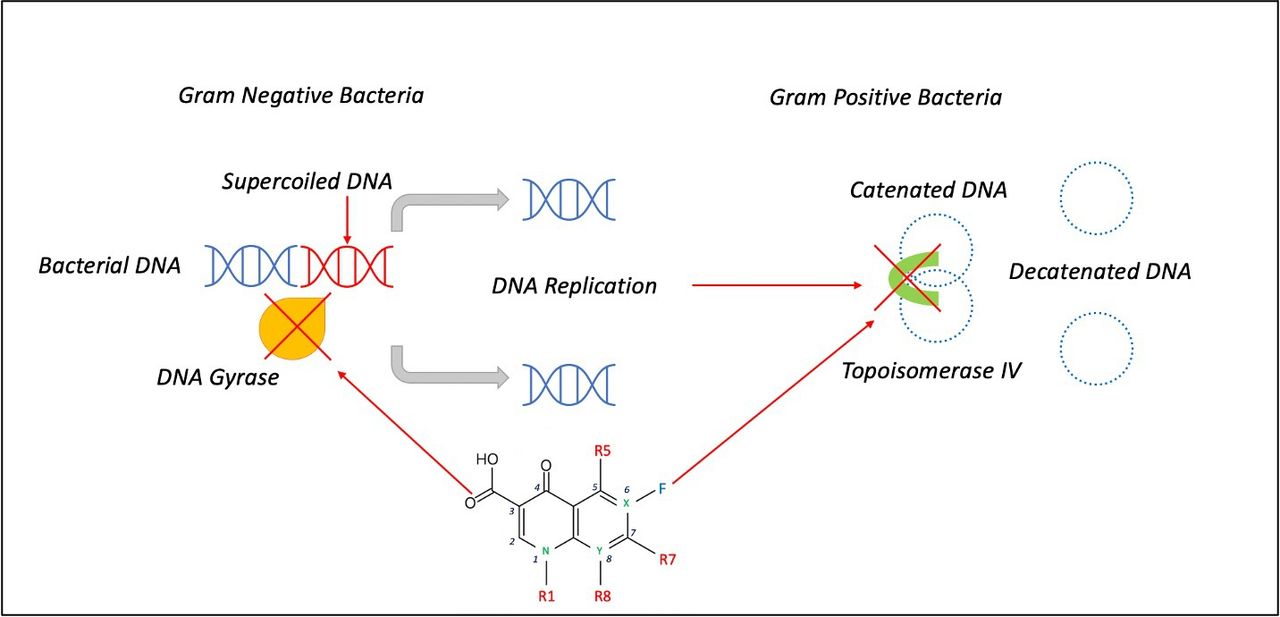
The FQs are bactericidal drugs. They kill bacteria by binding to and inhibiting the ligase activity of the bacterial type 2A topoisomerase enzymes, DNA gyrase and type IV topoisomerase, that are essential for bacterial DNA synthesis (figure 5).9 Although they target Gram-negative organisms predominantly through activity against gyrase, and Gram-positive bacteria through inhibition of topoisomerase IV, there are exceptions to this rule.9 Each topoisomerase exists as a heterotetramer; gyrase is composed of paired GyrA and GyrB subunits and topoisomerase IV of paired ParC and ParE subunits. Both topoisomerases catalyse double-stranded breaks in the bacterial DNA, insert another DNA fragment, and close the loop. These complexes, therefore, not only inhibit the DNA replication fork but also promote double-strand breaks, resulting in bacterial cell death. The human topoisomerases are fused as homodimers, and they are not susceptible to the action of FQs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mechanism of action of fluoroquinolones. DNA gyrase blockade inhibits the supercoiling of bacterial DNA in gram-negative bacteria, while topoisomerase IV inhibition prevents the segregation of replicated DNA in gram-positive bacteria. Adapted from Rusu et al. 42
Mechanisms of FQ resistance
There are three routes to acquired FQ resistance.
Chromosomal mutations
Single amino acid substitutions in either gyrase or topoisomerase IV can lead to AMR by altering the topoisomerase target enzyme and inhibiting FQ binding to the DNA cleavage complex.82 Resistance mutations most commonly occur in amino-terminal domains within GyrA of gyrase or ParC of topoisomerase IV, termed the quinolone resistance determining regions.83 For example, the most common site of mutation in the GyrA gene in E. coli is at the Ser83 amino acid residue, accounting for 90% of resistant isolates, followed by the Asp87 residue. Single target mutations can result in an eight to sixteen-fold increase in resistance.82 First-generation FQs have a single target and are particularly vulnerable to this resistance mechanism. In contrast, fourth-generation FQs with similar activities against topoisomerases II and IV require mutations in both topoisomerases. For example, besifloxacin has a dual inhibitory activity against DNA gyrase and topoisomerase IV than ciprofloxacin and moxifloxacin and is less affected by target enzyme mutations.
Altered drug permeation
Acquired FQ resistance by altered drug permeation occurs mainly by mutations in genes encoding regulatory proteins that control the transcription of efflux pumps or porins. In Gram-positive bacteria, it is predominantly overexpression of efflux pumps that lead to a loss of FQs from the cytoplasm.82–84 For example, increased expression of efflux pumps NorA,42, 43 NorB,44 and NorC45 are associated with a four to eightfold increase in FQ resistance in S. Aureus.84 Most efflux pumps have many substrates, and overexpression generally links FQ resistance to multidrug resistance. In Gram-negative bacteria, a reduction in porins, through which FQs access the periplasmic space, contributes to resistance.82–84
Plasmid-mediated resistance
Plasmids can pass FQ resistance genes between bacteria by horizontal gene transfer. Three genes encoded by the extrachromosomal bacterial plasmid, known as plasmid-mediated quinolone-resistance genes, promote FQ resistance. The bacterial Qnr gene encodes a protein with a pentapeptide motif that binds to the topoisomerases and blocks the interaction with FQs. Expression of Qnr has proven to result in a sixteen-fold increase in MIC of ciprofloxacin to E. Coli J53.83 85 Aminoglycoside acetyltransferase is capable of FQ degradation mediated by acetylation. The oqxAB and qepA genes encode efflux systems, promoting FQ removal from the bacterial cell.
Overuse and veterinary use of FQs may be a major cause for increased resistance, which has made some types of systemic infections difficult to treat. Although there is little evidence that using a topical FQ for bacterial keratitis is a strong driver for the absolute loss of bacterial susceptibility, antimicrobial stewardship and monitoring for levels of resistance is nevertheless important.12 24 In ocular isolates there is emerging FQ resistance to Gram-positive bacteria, particularly Streptococcus spp and Staphylococci, and multidrug resistance in MRSA. Maintaining the concentration of an antimicrobial agent above the MIC of the least susceptible, single-step bacterial mutants (the mutant prevention concentration) can help to reduce the selection of resistant clones.86 A high Cmax value with a low MIC is ideal. A Cmax:MIC ratio of >10 reduces the selection of resistant clones, while a ratio of <4 is suboptimal.46 In MK, the expected concentration in the cornea is likely to exceed a ratio of 10; however, there are variations of the reported cornea and aqueous concentrations of several FQs. Because the lower quartile of the reported corneal concentrations may be close to the MIC, the Cmax:MIC ratio may be closer to 1 than 10. For example, the MIC90 for Moxifloxacin against P. aeruginosa and S. aureus keratitis isolates is 1–4 mg/L while the lower quartile concentration in the cornea is approximately 6 mg/L (ie, the Cmax in 25% of cases) the Cmax:MIC ratio will vary in 25% of cases between 1.5 to 6. Intuitively, the frequency of administration (dose) should also not be reduced to a stage where there is the risk of a low Cmax:MIC, such as with BD drop administration for prolonged periods after treatment of the acute episode. Prolonged entry of a topically administered agent to the nasopharynx and gastrointestinal tract could expose endogenous bacteria to an antimicrobial at a low concentration, increasing the risk of resistance in these sites. In general, antimicrobials which target both gyrase and topoisomerase IV, for example, fourth and fifth generation FQs, are less susceptible to AMR.
Conclusions
We present data on the suitability for ophthalmic use of both the licensed and unlicensed FQs. Although the evidence is incomplete, FQs have good tissue cornea penetration and would thus achieve MIC90 levels for most of the common pathogens for bacterial keratitis. There is, however, significant variability in the reported corneal and aqueous concentrations, so relying on the reported mean concentration is inappropriate. The first quartile may be a more reliable value. In addition, there is little information on the biological activity of the FQs in the cornea and aqueous. The information presented in online supplemental table 1 and tables 1, 3 and 4, in particular the MIC90, could aid the clinician when presented with the MIC of the isolated bacteria to decide to continue the prescribed FQ at an increased dosage (concentration and frequency of application) or route of application, change to another FQ (licensed or unlicensed), or add a second antimicrobial to achieve an additive or synergistic effect. Routine use of susceptibility testing predicts the likelihood of treatment success and helps identify resistant strains. Ophthalmic breakpoints should be established to guide the clinician. Because these are unlikely to be available soon, microbiology laboratories need to provide the clinician with the MICs of isolates to available antimicrobials. Further investigation of potential options for combination therapy based on susceptibility testing may improve clinical outcomes while minimising the development of resistance.
Ethics statements
Patient consent for publication
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors contributed to the manuscript: helped with the writing and drafting and final document.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.