Article Text

Abstract
Objective Autosomal dominant vitreoretinochoroidopathy (ADVIRC) is associated with pathogenic variants in BEST1, which typically causes visual impairment in the late stage of disease. We present a pedigree with variable expressivity and the youngest case in the literature with visual impairment in early childhood.
Methods and analysis This is a retrospective, observational, case series describing multigenerational members of one family affected with ADVIRC. Patients underwent examination, ultra-widefield fundus photography and angiography, optical coherence tomography, full-field electroretinography (ffERG) and full-field perimetry.
Results Three affected members of the pedigree, one from each successive generation, were found to harbour a mutation, c.715G>A:p.Val239Met, in BEST1. The proband characterised in this report is, to our knowledge, the youngest documented case of ADVIRC in early childhood. Yet, this patient has the most severe retinal dysfunction compared with the father and paternal grandmother, whom exhibit classic characteristics of ADVIRC. Longitudinal data from the paternal grandmother showed that there was a rapid decline in ffERG responses (photopic decline worse than scotopic) from the fourth to fifth decade of life, which correlated with severe concentric constriction of visual fields.
Conclusion This multigenerational case series provides new insights into the ADVIRC disease spectrum and rate of progression. While ADVIRC typically causes a slowly progressive disease, we show that variable phenotypic expressivity is possible among affected members of the same family with the same mutation in BEST1. Thus, ADVIRC must also be considered in the differential diagnosis of paediatric patients with severe retinal dystrophy in early childhood.
- genetics
- imaging
- retina
- choroid
- electrophysiology
Data availability statement
Data are available upon reasonable request. Data availability statement: Dare available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Autosomal dominant vitreoretinochoroidopathy (ADVIRC) is associated with pathogenic variants in BEST1, which typically causes visual impairment in the late stage of the disease.
What are the new findings?
We describe the youngest case of ADVIRC in the literature with visual impairment in early childhood.
How might these results change the focus of research or clinical practice?
ADVIRC must be considered in the differential diagnosis of paediatric patients with retinal dystrophy in early childhood.
Introduction
Autosomal dominant vitreoretinochoroidopathy (ADVIRC, OMIM#193220) is an ultra-rare hereditary dystrophy first described by Kaufman et al in 19821 and linked pathological variants in BEST1.2 BEST1 (*607854) is located on chromosome 11q12.3 and encodes BEST1 (bestrophin 1) a 68 kD protein3 4 that has been proposed to act as an ion channel in the basolateral plasma membrane of the retinal pigment epithelium (RPE) and regulating calcium homoeostasis.5–7 Mutations in BEST1 are associated with a spectrum of phenotypes and has been described by at least five distinct presentations: Best vitelliform macular dystrophy (#153700), adult-onset foveomacular vitelliform dystrophy (#153700), autosomal recessive bestrophinopathy (#611809), retinitis pigmentosa (#613194) and ADVIRC.5 6 It should be noted that microcornea, rod-cone dystrophy, cataract and posterior staphyloma as described by Reddy et al in 20038 is likely the same disease as ADVIRC.5 More than 270 variants in the BEST1 gene have been reported (Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk/ac/gene.php?gene=BEST1), but the exact physiopathology of how different variants in this gene can cause distinct clinical phenotypes remains unclear.7
The hallmark of ADVIRC is a circumferential chorioretinal degeneration in the periphery with hyperpigmentation and a well delineated posterior boundary.9 Other features include nanophthalmos, microcornea, early cataract formation, angle closure glaucoma, fibrillar condensation of the vitreous, retinovascular abnormalities10 and chorioretinal macular atrophy as a late stage of the disease.11 12 Unlike Best vitelliform macular dystrophy, adult-onset vitelliform macular dystrophy and autosomal recessive bestrophinopathy that primarily affect the macula, patients with ADVIRC typically have a normal appearing macula.7 However, over time, patients with ADVIRC can present with cystoid macular oedema, epiretinal membrane, chorioretinal macular atrophy and cone dysfunction.12–14 Only five missense variants, p.Val86Met, p.Val239Met, p.Tyr236Cys, p.Val235Ala7 and p.Gly83Asp11 have been associated with ADVIRC. Nachtigal et al demonstrated that these five variants increased anion permeability in RPE cells.6
Herein multimodal images of three members of an affected three-generation family with the known p.Val239Met in BEST1 are described. We present the youngest patient in the literature with a clinical and molecular diagnosis of ADVIRC, who is also the most severe case within the family.
Materials and methods
This study was performed in accordance to the Declaration of Helsinki and protection of the patient’s identity. All subjects were provided with written informed consent for the use of personal medical data for scientific purposes and publication.
Phenotype description
Best-corrected visual acuities (BCVA), slit-lamp examinations, fundus examinations, intraocular pressures were extracted from chart reviews. Ultra-widefield retinal imaging (Optos California, Marlborough, Massachusetts, USA), fundus autofluorescence (FAF) (Optos California), optical coherence tomography (OCT) (Spectralis, Heidelberg Engineering, Heidelberg, Germany) were obtained using standard clinical protocols. Only the proband underwent a sedated OCT (Leica/Bioptigen, Leica Microsystem, Wetzlar, Germany). All patients underwent full-field electroretinography (ffERG) (Custom, Portland, Oregon, USA)15 in accordance to the International Society for Clinical Electrophysiology of Vision.16 The father and grandmother underwent kinetic visual field (Goldmann perimetry or Octopus 101 or Octopus 900; Haag-Streit AG, Köniz, Switzerland), and fluorescein angiography (FA) (Optos California). The grandmother also underwent non-widefield colour pictures and FA (Zeiss FF450 camera, Carl Zeiss Meditec AG, Jena, Germany).
Genetic testing
Genetic testing was performed for the proband at the Baylor College of Medicine Human Genome Sequencing Center (Houston, Texas, USA) via next generation sequencing with copy number variation (CNV) analysis of 266 genes associated with inherited retinal conditions. The panel targeted protein coding exons, exon–intron boundaries (±20 base pairs) and selected non-coding, deep intronic variations. Genetic counselling was provided before and after genetic testing. Variants identified were subsequently validated by Sanger sequencing.
The proband’s father and paternal grandmother had genetic testing completed via next generation sequencing (Baylor College of Medicine Human Genome Sequencing Center). It did not include CNV analysis. It included analysis of over 250 genes associated with inherited retinal conditions. Pretest and post-test genetic counselling occurred.
Results
Proband (V-3)
The female proband (figure 1) presented at 20 months of age to the Ophthalmic Genetics Division at the Casey Eye Institute. The family reported she was bumping into and tripping over objects on her sides, tripping over things and experiencing difficulty with night vision. Visual acuity measurements demonstrated that she could fix and follow. In both eyes, anterior segment examination was unremarkable, but fundus examination revealed disc pallor, attenuated vessels, a blunted macular reflex and diffuse RPE atrophy with coarse pigment mottling and visible choroidal vessels (figure 2A). On FAF, there was relative hyperautofluorescence of the posterior pole and hypoautofluorescence of the mid and far periphery (figure 2B). There was outer retinal attenuation beyond the macula with foveal hypoplasia as evidenced by retained inner retinal layers in both eyes on OCT (figure 2C). Sedated ffERG showed a pattern of severe rod-cone dysfunction (figure 2D). At her most recent visit at 3 years of age, BCVA was 20/40 in the right eye and 20/50 in the left eye with a refraction of plano +2.00×90° and plano +2.50×85°, respectively. Due to severe tunnel vision, she was undergoing cane training for navigation.

Pedigree. Family history is positive for the proband’s father and paternal grandmother with a diagnosis of ADVIRC and familial variant in the BEST1 gene, both of whom have been previously seen in the same ophthalmic genetics clinic. No significant vision concerns for the proband’s two older sisters, they have been followed by a general paediatric ophthalmology clinic. Of note, there are other paternal family members with unexplained vision loss, to our knowledge they have not had a thorough workup. Legend: Blue represents individuals with a known diagnosis of ADVIRC. Red represents individuals with unexplained vision loss, unknown diagnosis. ADVIRC, autosomal dominant vitreoretinochoroidopathy; BEST1, bestrophin 1; ARMD, age-related macular degeneration.
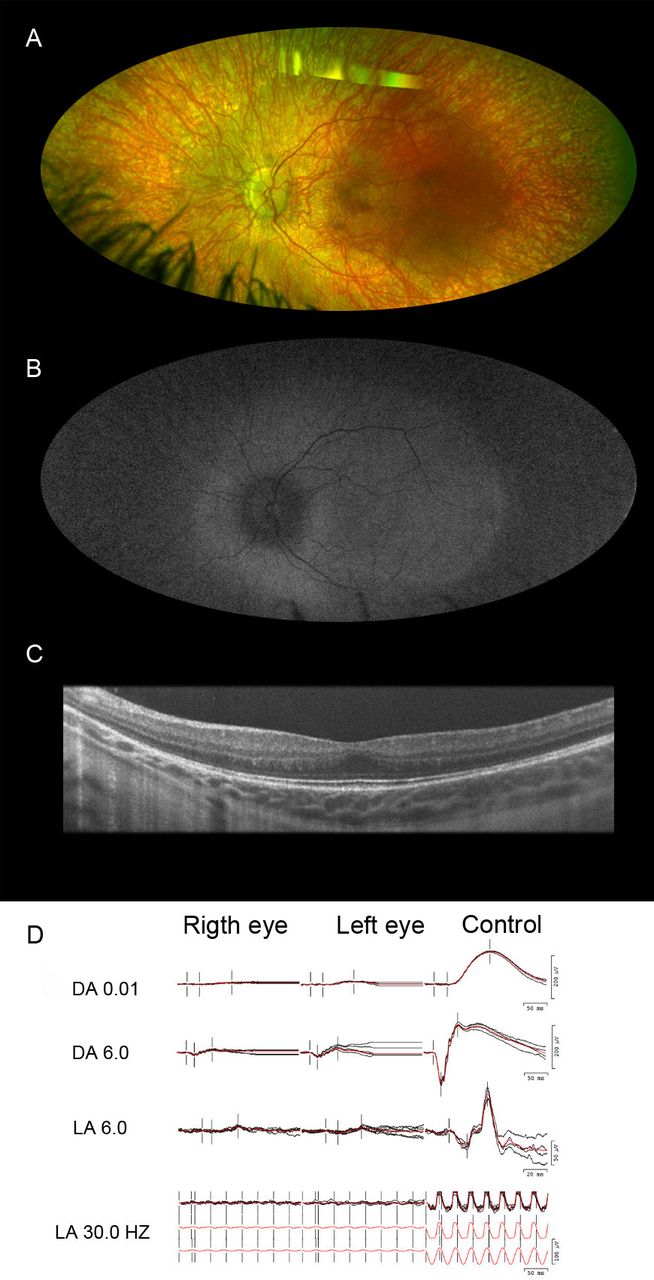
Multimodal ophthalmic imaging of the proband with autosomal dominant vitreoretinochoroidopathy and a pathogenic variant p.Val239Met in bestrophin 1 gene. (A) Fundus image of the left eye shows disc pallor, attenuated vessels, mottled macula and coarse mottling atrophy in the mid periphery and periphery with hyperpigmented spots distributed throughout the peripheral retina. Note the visibility of large choroidal vessels. (B) Fundus autofluorescence imaging of the left eye shows a peripheric well-demarcated circumferential hypoautofluorescence. (C) The horizontal line scan from the optical coherence tomography of the fovea in the left eye demonstrates retained inner retina layer causing foveal hypoplasia. (D) Sedated full-field electroretinography of both eyes demonstrates severe rod-cone dysfunction. DA 0.01, scotopic dim; DA 6.0, scotopic bright; LA 6.0, photopic single; LA 30.0 Hz, photopic 30 Hz.
Father (IV-2)
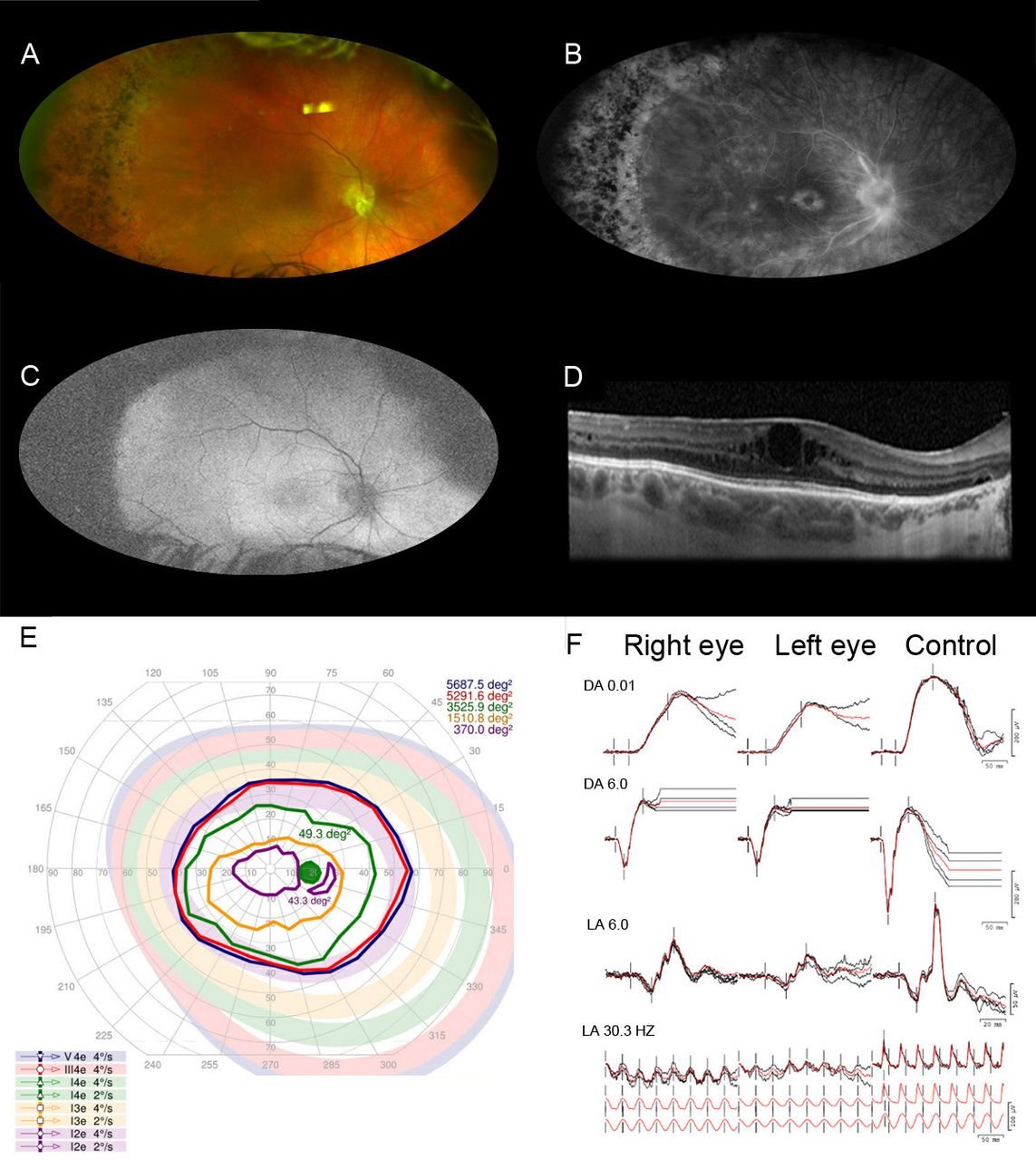
The proband’s father presented at 39-year-old with reports of decreasing peripheral vision and a history of progressive nyctalopia and light sensitivity since age 15. He had cataract surgery in both eyes at 31 years of age and a history of angle-closure glaucoma. His BCVA was 20/50 in the right and 20/70 in the left eye. Prior to cataract surgery, his refraction was −0.25+1.25×30° and −0.50+1.75×135°, respectively, in the right and left eye. His intraocular pressures were normal while on dorzolamide two times per day in both eyes. The anterior segment examination revealed microcornea with a diameter of 9.5 mm in both eyes. Although the anterior chambers appeared deep without peripheral iridotomies, gonioscopy was not performed. The fundus examination revealed bilateral disc pallor with prepapillary fibrous tissue, blunted macular reflex with pigmentary mottling, vascular attenuation and a well-demarcated region of retinal atrophy and pigment changes 360 degrees in the retinal periphery (figure 3A). The FA showed window defects in the periphery with a few scattered chorioretinal hypofluorescent spots in both eyes (figure 3B). The autofluorescence showed bilateral diffuse hyperautofluorescence in the posterior pole and peripheral hypoautofluorescence, that was consistent with RPE atrophy (figure 3C). The OCT images showed cystoid macular oedema and epiretinal membrane in both eyes (figure 3D). Kinetic perimetry demonstrated constriction of the V4e isopters down to about 100 degrees horizontal diameter in each eye (figure 3E). FfERG showed a generalised retinal dysfunction with a pattern of a moderate cone and rod dysfunction (figure 3F). For the management of cystoid macular oedema, ketorolac was prescribed; however, he was lost to follow-up.

Multimodal ophthalmic imaging of the father of the proband with autosomal dominant vitreoretinochoroidopathy (p.Val239Met in BEST1). (A) Fundus photography of the right eye showing disc pallor with glial material peripapillary, and inferior, blunted macula with pigmentary changes, vascular attenuation and a sharp classical between a region of normal retina and a region of pigmented changes and yellowish deposits in the periphery 360 degrees. (B) The fluorescein angiography shows early proximal arterial hyperfluorescence with late perivascular staining and late cystoid macular oedema and scattered few chorioretinal hypofluorescent spots in the periphery. (C) Fundus autofluorescence of right eye shows circumferential peripheric hypoautofluorescence and an irregular hyperautofluorescence in posterior pole. (D) The horizontal line scan from the optical coherence tomography of the macula in the right eye reveals cystoid macular oedema and epiretinal membrane. (E) Kinetic visual field of the right eye demonstrates constriction of peripheral isopters down to 100 degrees central island to V4e. (F) Full-field electroretinography shows a generalised retinal dysfunction with a pattern of a moderate cone-rod dysfunction. DA 0.01, scotopic dim; DA 6.0, scotopic bright; LA 6.0, photopic single; LA 30.0 Hz, photopic 30 Hz.
Paternal grandmother (III-2)
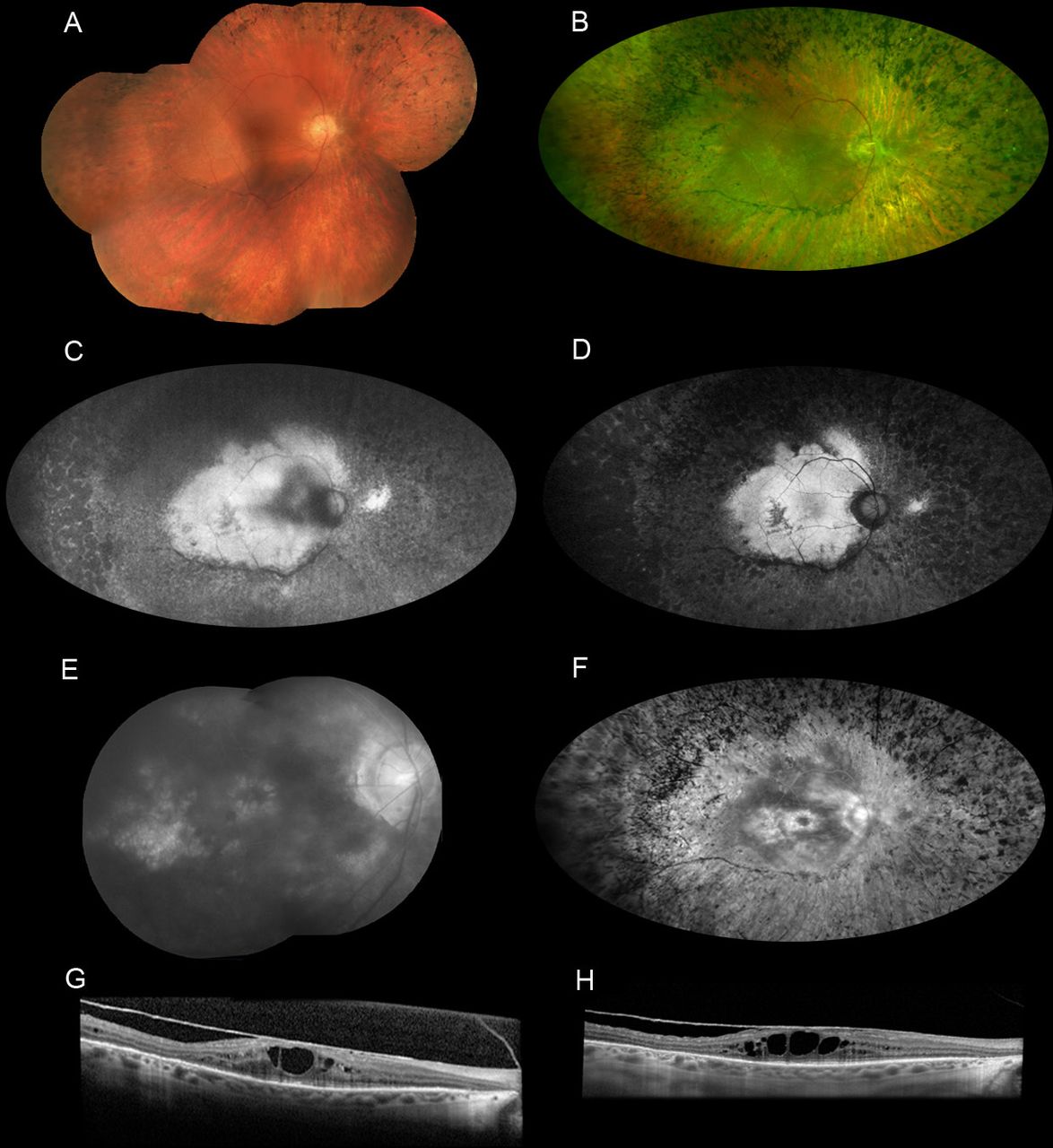
The paternal grandmother is a 59-year-old woman who has been followed in our clinic for the past 30 years. She was previously diagnosed with autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV) until she was linked with the family pedigree and genetic testing was performed. She reported progressive difficulty seeing at night since age 20–30 with constriction of peripheral vision thereafter. The patient had always been extremely sensitive to light and glare. Her BCVA on record was 20/50 in both eyes at 48 years old with a refraction of plano in the right eye and +0.75 sphere in the left eye. However, her acuity declined gradually over the subsequent decade. She underwent cataract surgery in both eyes when she was 57–58 years old. Her BCVA after cataract surgery was 20/200 in the right eye and 20/125 in the left eye with a refraction of −1.75+2.50×70° and −1.00 sphere, respectively. The anterior segment examination was significant for a small corneal diameter of 10.5–10.6 mm and gonioscopy revealed closed angles in two of four quadrants in both eyes. In addition, the axial length was 20.6 mm in the right eye and 20.4 mm in the left eye, which is consistent with nanophthalmos. The fundus examination revealed diffuse pigmentary changes and pigment clumping. Atrophy of retinal pigment epithelium, and cystoid macular oedema, increased over the years as shown on multimodal ophthalmic imaging from different ages (figure 4A–H) . Vitreomacular traction and epiretinal membranes were also evident on OCT. These anatomical changes were correlated with function loss on perimetry and electrophysiology (figure 5A–D). Over three decades, her visual fields progressively constricted (figure 5A–C), and the ffERG showed progressively rod-cone dysfunction (figure 5D). At 34 years of age, her ffERG showed only a mild–moderate rod-cone dysfunction. Fourteen years later, there was severe dysfunction of both rods and cones, with prolonged rod and cone implicit times.

Multimodal ophthalmic imaging of the paternal grandmother with autosomal dominant vitreoretinochoroidopathy (p.Val239Met in BEST1). All pictures in the left at 59-year-old compared with old pictures in the right. (A) Fundal photomontage (FF450 Zeiss) of the right eye at 49-year-old showing the mild and far peripheral pigmentary changes. (B) Widefield photograph (Optos California) at 59-year-old of right eye demonstrates clear media, waxy disc pallor, attenuated vessels with associated pigment clumping and bone spicules in the mid periphery and periphery that increased compared with 10 years ago. (C) Fundus autofluorescence (FAF) of the right eye at 54-year-old compared with FAF (D) at 59-year-old showing irregular area of central hyperautofluorescence and a circumferential hypoautofluorescence surrounding the disc and temporal arcades in a centripetal progression of the hypoautofluorescence beyond posterior pole. Expanded pictures of fluorescein angiogram (FA) at 49-year-old (E) showing petaloid macular hyperfluorescence in late phase due to staining and oedema and a hyperfluorescence due to a window defect temporal to the fovea area. FA repeated at 59-year-old (F) showing the enlargement of the foveal hyperfluorescence due to increased macular oedema as well as increased diffuse hyperfluorescence due to window defect and decreased relative hypofluorescence of the residually intact retina in the posterior pole. The attenuated vessels are not perfused early or late during the examination. There is also a hypofluorescent bone-spicule pattern in the periphery. (G) The horizontal line scan from the optical coherence tomography (OCT) of the macula in the right eye at 49-year-old reveals cystoid macular oedema and epiretinal membrane (ERM). OCT at 59-year-old (H) showing sustained ERM and increased macular oedema.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Functional tests over decades. (A) The first Goldmann perimetry was done in her 30s. (B) The octopus 101 was done in her 40s. (C) Octopus 900 was done in her 50s. All isopters to all targets decreased over years remaining less than 40 continuous horizontal degrees to V4e in both eyes in her last kinetic visual field. (D) The response amplitudes of three different full-field electroretinography over two decades were compared into a data base of age-matched controls. Amplitudes were the average of the right and left eye responses for all measurements and are presented as a percentage of the lower limit of age-matched normal response. All amplitudes decreased to minimal responses in both scotopic and photopic revealing a progression of the disease and severe retinal rod and cone dysfunction in both eyes in late-stage autosomal dominant vitreoretinochoroidopathy.
Genotype description
Genetic testing of the proband, her father and paternal grandmother detected a heterozygous missense pathogenic variant in the BEST1 gene c.715G>A:p.Val239Met, which has been reported previously.2 The valine residue is highly conserved across species and there is a small physiochemical difference between Val and Met. It is predicted to be damaging by all in silico tools used (Polyphen, Sift, Muttaster). The proband was also found to have one likely pathogenic variation in WDR19 (c.3484–2A>C) and RDH11 (729dup:p.Ser244Ilefs*34), and one variation of uncertain significance in SPATA7 (c.730A>G:p.Thr244Ala) and TULP1 (c.616G>A:p.Asp206Asn). These results were not clinically significant given that these genes are associated with recessive retinal dystrophies. Segregation studies were not completed for these variants.
Discussion
This study describes a three-generation family with a molecular and clinical diagnosis of ADVIRC. The pedigree is consistent with an autosomal dominant inheritance pattern, which is associated with a previously reported pathogenic variant in BEST1, namely c.715G>A (p.Val239Met).2 The affected members of this family exhibited the typical retinal features of ADVIRC with well-demarcated circumferential peripheral pigment changes, vitreous membranes/opacities and chorioretinal atrophy. In addition to retinal dystrophy, our patients also had signs of developmental ocular abnormalities, such as nanophthalmos, microcornea, closed angle glaucoma and early-onset cataract, which is consistent with prior reports.2 Yardley et al reported that the V239M mutation in BEST1 is associated with a more severe phenotype, including rod-cone dystrophy and posterior staphyloma. While we did not appreciate staphyloma in this family, there was a spectrum of rod-cone dysfunction, with the youngest affected family member having the most severely attenuated ffERG responses. However, the proband’s OCT did not demonstrate cystoid macular oedema, which is expected as a late macular involvement as seen in the father and grandmother. In addition, the maternal grandmother exhibited a severe decline in ffERG responses from the fourth to fifth decade of life, which was correlated with an equally rapid decline in visual field during the same time period.
To our knowledge, we have also characterised the youngest patient with ADVIRC reported to date in the literature. Given the rarity of the disease, there are very few published cases of affected children. In a cross-sectional study of 12 patients between the ages of 11 and 64, decreases in the ffERG correlated with advanced age.17 In contrast, the ffERG in our 20 months old proband was much more attenuated than her father. Most studies have reported normal visual acuity the first few decades of life, before progressing to visual impairment in later age.11 14 18 However, the visual behaviour in our affected proband was consistent with decreased vision in early childhood, which is indicative of a more severe phenotype than her father and paternal grandmother who had onset of symptoms at 15 and 20–30 years of age, respectively. These observations not only support the notion that this mutation in BEST1 can be associated with a severe phenotype, but also with variable expressivity within the same family.
The range of disease severity observed in our family is not surprising given that diseases caused by pathogenic variants in the bestrophin gene have high interfamilial and intrafamilial clinical variability such as the age of onset, the disease progression and visual impairment6 19 20 even among individuals with the same BEST1 variant.21 22 The aetiology of the variable expressivity and penetrance remains to be elucidated, however environmental factors and/or variants or polymorphisms in other genes interacting with the BEST1 gene may play a role.21–24 Esumi and colleagues demonstrated that transcription factors MITF, OTX2 and possibly CRX, may act as modifiers of BEST1 expression,20 25–27 although these mechanisms are still poorly understood.28 In addition, mutations in PRPH2, IMPG1 and IMPG2 can overlap phenotypically with BEST1, which suggest there may be some common pathway in the pathophysiology that is yet to be elucidated. The ADVIRC phenotype is hypothesised to be a result of aberrant splicing of BEST1. Using a minigene assay in HEGK293 cells, Yardley et al demonstrated that their missense pathogenic variants (p.Tyr236Cys, p.Val86Met and p.Val239Met) in BEST1 disrupts splicing and results in exon skipping and an in-frame deletion.2 However, Chen and colleagues studying a novel missense mutation, p.Gly83Asp, using the same minigene system failed to show any effect on splicing.11 In addition, there has been conflicting studies showing whether variants, p.Val235Ala or p.Val86Met, may affect pre-messenger ribonucleic acid (mRNA) splicing.6 7 29 Further studies are needed to enhance our understanding of the pathophysiology of BEST1 mutations as it relates to expressivity, penetrance and the phenotypic spectrum of BEST1-related retinopathies.
As highlighted by the initial clinical diagnosis of the grandmother, the differential diagnosis of ADVIRC in advanced cases, includes ADNIV,30 which is an inherited autoimmune disease due to mutations in the calpain 5 gene located on chromosome 11q13.5.31 Cases of ADNIV are marked by severe intraocular inflammation and retinal degeneration.30–32 The common features of these two autosomal dominant disorders, ADVIRC and ADNIV, are the inheritance pattern, cells in the vitreous, cystoid macular oedema, spots of peripheral retinal pigmentation and risk of retinal neovascularisation.30 However, the hallmark of ADVIRC in early to moderate disease is a sharp boundary between the normal and abnormal retina, which is not seen in ADNIV.30 In addition, the ffERG of patients with ADNIV typically have a disproportionate reduction in the b-wave amplitude in early diseases, but the ffERG responses can be diffusely extinguished in severe cases for both ADNIV and ADVIRC. These overlapping signs and symptoms underscore the need for genetic testing, especially in advanced disease.
In summary, ADVIRC is a slowly progressive retinal degenerative disorder with variable expressivity due to mutations in BEST1, which is associated with a wide spectrum of retinal disorders. Given the variable presentation and overlapping features with other degenerations, genetic testing is an essential diagnostic tool, especially in advanced disease. Follow-up is essential to monitor for and manage comorbidities. While there is currently no cure for BEST1-associated retinopathies, there is hope that ongoing work will eventually lead to the development of genetic treatment.33 34
Data availability statement
Data are available upon reasonable request. Data availability statement: Dare available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Research Ethics Committee of Oregon Health and Science University—IRB ID 00002735 approved this study. A signed BMJ patient consent form was obtained directly from patients.
Acknowledgments
The authors thank the family members who consented to participate in this study.
References
Footnotes
Contributors Conceptualisation, MEP, PY; methodology, PY; data curation, MMP, MEV, AB, RC, RGW, PY; writing-original draft preparation, MMP, MEV, AB, PY; writing—review and editing, MMP, MEV, AB, RC, MEP, RGW, PY; visualisation, MEP, PY; supervision PY. All authors have read and agree to the published version of the manuscript. PY accepts full responsibility for the work and/or the conduct of the study, had access to the data, and controlled the decision to publish.
Funding MMP, this study was financed in part by the Coordenação de Aperfeiçoamento de Pessoa de Nível Superior – Brazil (Capes) – Finance Code 001; RC, NIH NEI R01EY022356 and R01EY018571; PY, NIH K08EY026650, Foundation Fighting Blindness Career Development Award CD-NMT-0714–0648; MEP, grants from Foundation Fighting Blindness; other from Allergan/Editas, other from Spark Therapeutics, other from Wave Biosciences, other from Astellas Pharmaceuticals, other from RegenexBio, other from Iveric, other from Biogen, other from Novartis, other from Adverum, other from Gensight, other from ProQR, other from Horama, other from Eyevensys, other from Nayan, personal fees and other from Nacuity, personal fees and other from Ocugen, personal fees and other from Verede, other from Sparing Vision, other from AGTC, other from Sanofi, outside the submitted work. ‘Supported by grant P30 EY010572 from the National Institutes of Health (Bethseda, MD), and by unrestricted departmental funding from Research to Prevent Blindness (New York, NY).’
Competing interests RGW, Oregon Health & Science University, in the name of RGW, holds US patent no. 8657446, Method and apparatus for visual field monitoring, also known as Visual Field Modelling and Analysis or VFMA (P), which is licensed but no royalties have yet accrued to RGW. RGW is on advisory boards for AGTC, Janssen Research and the Foundation Fighting Blindness (F).
Provenance and peer review Not commissioned; externally peer reviewed.