Article Text

Abstract
Age-related macular degeneration (AMD) is the most common eye disease in elderly patients, which could lead to irreversible vision loss and blindness. Increasing evidence indicates that amyloid β-peptide (Aβ) might be associated with the pathogenesis of AMD. In this review, we would like to summarise the current findings in this field. The literature search was done from 1995 to Feb, 2021 with following keywords, ‘Amyloid β-peptide and age-related macular degeneration’, ‘Inflammation and age-related macular degeneration’, ‘Angiogenesis and age-related macular degeneration’, ‘Actin cytoskeleton and amyloid β-peptide’, ‘Mitochondrial dysfunction and amyloid β-peptide’, ‘Ribosomal dysregulation and amyloid β-peptide’ using search engines Pubmed, Google Scholar and Web of Science. Aβ congregates in subretinal drusen of patients with AMD and participates in the pathogenesis of AMD through enhancing inflammatory activity, inducing mitochondrial dysfunction, altering ribosomal function, regulating the lysosomal pathway, affecting RNA splicing, modulating angiogenesis and modifying cell structure in AMD. The methods targeting Aβ are shown to inhibit inflammatory signalling pathway and restore the function of retinal pigment epithelium cells and photoreceptor cells in the subretinal region. Targeting Aβ may provide a novel therapeutic strategy for AMD.
- macula
- angiogenesis
- inflammation
- retina
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Age-related macular degeneration (AMD) has been regarded as the leading cause of progressive central vision loss and blindness in the elder individuals due to the impairment of photoreceptor cells (PRCs) and retinal pigment epithelium (RPE) cells caused by the formation of drusen in Bruch’s membrane and the growth of leaky blood vessels beneath the retina.1–4 In the past 10 years, considerable attention has been paid to the significant role of oligomeric amyloid β-peptide (Aβ) in the developed pathogenesis of AMD. Aβ comprises peptides between 39 and 43 amino acid residues which are produced by the proteolytic cleavage process of amyloid precursor protein (APP) with the utilisation of multisubunit enzyme complex and membrane-bound aspartyl protease.5–7
Previous studies suggested that oligomeric Aβ has been crucially involved in the pathogenesis of Alzheimer’s disease (AD). Under conditions with Aβ, microglia could be stimulated and activated chronically, which leads to extensive neuronal apoptosis. Moreover, aggregation of Aβ could induce the dysfunction of microtubules associated protein-2, which successively disrupts the microtubules structure and process of axonal transport.8 Aβ can also disrupt the glucose metabolism in the brain by vying with insulin for binding to its receptor.9 Increasing evidence indicates that AD and AMD share similar pathophysiological features, such as neuroinflammation and oxidative stress.10 11 Aβ has been affirmed to be specifically prevalent in the extracellular soft drusen deposits of patients with AMD.12–14 Administration of Aβ in the subretinal region of C57BL/6 mice has shown similar pathology with AMD which exacerbates the senescence of RPE cells and retinal degeneration, suggesting that Aβ may be responsible for the characteristics of AMD.15 Recent studies have identified multiple different isoforms of Aβ, 40-residue peptide Aβ (1-40) and 42-residue peptide Aβ (1-42), which were the major constituents of drusen deposits in the subretinal space of patients with AMD.16 17 The overexpression of Aβ (40-residue and 42-residue) leads to the formation of drusen-like deposits in subretinal space in the eye and produces RPE atrophy.13 16 18 19 Moreover, senescent models with AMD present increasing amount of Aβ drusen in outer segments layer, leading to the outcome of PRCs loss and aberrant localisation of RPE cells.5 20 Several pathways involved in Aβ enhancing the formation of AMD have been clarified in current researches.
Aβ induces inflammasome in AMD
Inflammatory activity is a rapidly deteriorating mechanism and induced by several stress factors such as increased oxidative stress and decreased proteostasis, which has been tightly related to the pathogenesis of AMD.21 22 Previous evidence has shown that Aβ-induced inflammatory activity has a significant impact on the pathology of AMD and could be induced in distinct pathways.21 In the RPE cells, Aβ (1-40) and Aβ(1-42) can stimulate the expression of toll-like receptor 4 and Rel proteins and activate the nuclear factor-κB (NF-κB) signalling pathway.23 24 Activation of this pathway can upregulate the expression of proinflammatory cytokines, such as interleukin (IL)-6, tumour necrosis factor α (TNF-α), IL-1β, IL-18, which results in the event of nucleotide-binding oligomerisation domain-like receptors family pyrin domain containing 3 (NLRP3) inflammasome activity, and other apoptotic factors in the choroid and the neuroretina.23 25 26 Prolonged NLRP3 inflammasome activity induced by Aβ via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria-dependent reactive oxygen species (ROS) production leads to mitochondrial DNA damages, which eventually activates pyroptosis and apoptosis in RPE cells.27 28 Thus, the suppression of NLRP3 inflammasome activation may decrease the inflammation in RPE cells.29 Also, a new finding has illustrated that Aβ(1-42) can activate primary microglial, which results in the overproduction of proinflammatory cytokines, such as IL-1β and cyclooxygenase-2 (cox-2), and exacerbate the deterioration of visual function and PRCs apoptosis.30 Moreover, the activation of liver X receptor α (LXR α) and ATP binding cassette subfamily A member 1 (ABCA1) downregulates the expressions of proinflammatory cytokines, such as IL-6 and TNF-α, and suppresses the Aβ-induced inflammatory activity and senescent responses in RPE cells.31 Intravitreal injection of oligomeric Aβ suppresses the protein expressions of LXR α and ABCA1, which provokes inflammatory activity by upregulation of proinflammatory cytokines.32 Several medical agents, such as Brimonidine, Puerarin and Baicalin, have been shown to alleviate intracellular pyroptosis and viability damage through preventing Aβ-induced oxidative stress damage and inflammatory activity.33–35 Also, sirtuin 1 (SIRT1) has been identified as a protective factor, which suppresses the activation of NF-κB signalling pathway induced by Aβ in AMD.36 These results have shown the proinflammatory role of Aβ in AMD.
Aβ induces mitochondrial dysfunction in AMD
In AMD, Aβ dysregulates the level of mitochondria-associated proteins, such as pyruvate dehydrogenase and the electron transport chain complex IV, and disrupts the translocation of hydrogen from the matrix to the intermembrane space, which results in abnormal mitochondrial electrical activity and ROS increase.37 38 These aberrant activities could induce alterations in mitochondrial DNA and mitochondrial lipids, leading to mitochondrial impairment.37 38 Previous study has indicated that mitochondrial oxidative phosphorylation is inhibited by Aβ in AD.39 Recent study has shown mitochondrial impairment in AMD accompanied along with oxidative phosphorylation.40 The level of proteins related to the oxidative phosphorylation and mitochondrial dysfunction, such as Ndufs4 and Atp6v1g1, has been significantly downregulated in the initial stage of Aβ exposure and recovered after 24-hour exposure of Aβ in 661W cone PRCs.40 This mechanism is regulated by sirtuin signalling pathway, which has been shown to be involved in neuroprotection against toxicity in retina,41 and also tau protein, which is thought to be coexisted with Aβ42 and cause the suppression of oxidative phosphorylation.39 Tau protein in accumulation with Aβ induces the decline in oxidative phosphorylation in early stage of AMD.40 Moreover, under the presence of transcription factor PU.1/SPI1 induced by Aβ, NADPH oxidase activation occurs and induces the expression of NADPH oxidase (NOX) complex, such as NOX4-p22phox complex, leading to the outcome of mitochondrial dysfunction and excessive oxidative stress in RPE cells.43 Silencing of PU.1/SPI1 has been shown to impede the process of ROS production and mitochondrial dysfunction and protect the retinal structure and function from oxidative damage.43 This novel finding brings out new insight into preventing mitochondrial dysfunction induced by Aβ in RPE cells. From above, it is suggested that Aβ could lead to the consequence of mitochondrial dysfunction in AMD.
Aβ alters the function of ribosome in AMD
In initial stage of AMD, ribosomal protein synthesis has been aberrantly altered. Protein translation initiation factors, eIF2α, eIF3η and eIF5, and elongation factor, eEF2, are abnormally regulated, which induces ribosomal dysfunction in AMD.44 In recent study, ribosomal proteins are downregulated in the initial stage of the neurotoxicity induced by Aβ in AMD.40 Interestingly, after 24-hour treatment with Aβ, ribosomal proteins, such as Rpl29 and Rps19, are alleviated to its normal level in 661W cells.40 Meanwhile, it has been demonstrated that the differential effect of Aβ may be due to the recuperation of PRCs from oxidative stress.40 Thus, ribosomal proteins are negatively influenced in the early stage of the toxicity induced by Aβ treatment in AMD.
Aβ regulates the lysosomal pathway in AMD
Autophagy-lysosomal pathway has been elucidated to be a remarkable mechanism to prevent the accumulation of Aβ in the intracellular space in AMD.45 Previously, autophagy has been reported to be significantly involved in the process of Aβ clearance and degradation due to its property of clearing proteins. Successive autophagy activity could inhibit the accumulation of deleterious proteins, preventing the degeneration of RPE cells and decelerating the ageing process.46 In fact, Aβ could induce autophagy in RPE, which forms resistance to the formation of Aβ deposition, but the underlying molecular mechanisms are still unclear, which needs to be further explored.47 Previous findings have proved that autophagy-lysosomal system maintains retinal homeostasis and prevents retinal degeneration in AMD.48 Recent research has observed that lysosomal proteins are upregulated in the early stage of Aβ treatment, which protects cells from Aβ aggregation.40 However, after 24-hour Aβ exposure, lysosomal proteins are subtly downregulated, which indicates that Aβ might accumulate after long Aβ exposure.40 This has also been proved by a new research which found that Aβ accumulates in lysosomes under the circumstances of late-endocytic compartments and damages the function of RPE cells chronically.49 From this information, self-protection is induced through activating the autophagy-lysosomal pathway in the initial stage of Aβ exposure,40 rather than in the late stage.
Aβ affects RNA splicing in AMD
Spliceosome, a ribonucleoprotein (RNP) complex, comprising small nuclear RNAs (snRNA) and numerous proteins, has a significant role in the process of pre-mRNA splicing.50 Defects of mRNA processing and splicing are detected in AD according to previous research.51 Previous studies have also reported that the alterations in ubiquitous core snRNP proteins, pre-mRNA processing factor 3 (PRPF3), and splicing factor, retinitis pigmentosa 9, induce the aggregation of misfolded proteins, such as T494M mutant PRPF3, in PRCs, and lead to retinal degeneration.52 53 Recent finding has shown that, in the initial stage of Aβ treatment, several snRNP proteins reveal no change or subtle downregulation in PRCs. However, after 24-hour treatment with higher Aβ concentrations, proteins, such as Acin1 and Rbmx, are upregulated, which presents that aberrant alterations in RNA splicing is induced at a chronic exposure to Aβ.40 Thus, RNA splicing in AMD is abnormally influenced in late stage of Aβ exposure.40
Aβ modulates angiogenesis in AMD
Angiogenesis has been thought to be induced by the imbalance of angiogenesis-related factors.54 In AMD, vascular endothelial growth factor (VEGF) is highly expressed in subfoveal fibrovascular membrane, the surrounding tissue and the RPE cells, which stimulates the growth of new blood vessels in the subretinal region.55 Under the condition of low concentration of Aβ, RPE cells secrete significant level of pigment epithelium-derived factor (), an antiangiogenetic factor, through Aβ-receptor for advanced glycation endproducts pathway, which inhibits the apoptotic pathway leading to RPE cell growth.56 However, high concentration of Aβ treatment induces high expression of mRNA of VEGF in RPE cells and increases the death of RPE cells.56 Moreover, Aβ-induced mitochondrial ROS have been shown to promote the production of Aβ-stimulated angiogenic factor in ARPE-19 cells.57 These findings have shown that Aβ could regulate angiogenesis-related factors. Due to the accumulation of Aβ in AMD, it is speculated that Aβ could cause the growth of new blood vessels, which needs to be further explored.
Aβ alters actin cytoskeleton in AMD
Previous research has shown that Aβ can stimulate the loss of actin cytoskeleton integrity and function through abnormal phosphorylation events which induces microtubules instability and actin dynamics imbalance in AD.58 Recent findings suggest that accumulation of Aβ in intracellular region can also induce disorganisation of actin cytoskeleton and disruption of tight junction via NF-κB activation in AMD.40 59 Subretinally injected Aβ dislocates occludin and decreases the levels of occludin and zonula occludens‐1 mRNA expression in RPE cells.60 This molecular alteration destabilises the link between transmembrane and actin cytoskeleton and influences the transepithelial permeability of RPE cells.60 Recent findings have also shown that Tau, a microtubule-associated protein, is highly phosphorylated by the activation of glycogen synthase kinase 3 beta (GSK3β) in response to high concentration Aβ treatment, which upregulates the expression of cytoskeleton-associated proteins and induces reorganisation of cytoskeleton networks in PRCs.40 GSK3β inhibition has been observed in the early stages of Aβ exposure, which indicates the countering activity of the PRCs in response to Aβ-induced neurotoxicity.40 Moreover, keratins, intermediate filament proteins, are remarkably enriched in early timepoint and return to normal level after prolonged (24 hours) Aβ treatment in 661W cells, which suggests that keratin filaments are significantly influenced in early Aβ treatment.40 From above, it is suggested that Aβ could disorganise the cytoskeleton in RPE cells and PRCs in AMD.
Protective role of targeting Aβ in AMD
The approach to decrease or eliminate the accumulation of Aβ in the subretinal region has been imperatively developed in order to inhibit inflammatory activity and prevent visual loss efficiently. Evidence has shown that immunotherapeutic strategies, such as anti-Aβ antibodies treatment, could reduce neuronal damages in the retina and may recover the visual function.61 Moreover, anti-Aβ monoclonal antibody treatment can lead to the reduction of Aβ deposition and deactivates the plasma proteins in the complement system, which prevents or reverses the loss of eye vision.61 62 Previous research has reported that the mutation and loss-of-function of the triggering receptor expressed in myeloid/microglial cells-2 (TREM2) could negatively impact the efficiency of Aβ clearance.63 However, with incubating anti-miRNA-34a, TREM2 can be restored back to the normal homeostatic level, which recovers the ability to eliminate Aβ.63 Moreover, when NF-κB pathway induces and upregulates the expression of miRNA-34a, downregulation of TREM2 expression has been observed in human AMD.63 Thus, the discovered finding of NF-κB regulated, miRNA-34a-mediated TREM2 sensor-receptor circuit give novel thought about the utilisation of anti-NF-κB and anti-miRNA-based therapeutic strategies to clear Aβ deposits.63 Moreover, previous findings have shown that the possible reason for increasing amount of Aβ in the senescent models is the curtailment in the expression of neprilysin level and the growing activity of beta-secretase-1 leading to the higher level of Aβ synthesis in the RPE cells.64 65 Recent research has shown that intravitreal injections of neprilysin in mice model with AMD have shown the reduction of Aβ accumulation, which represents a potential method to slow down the development of AMD pathogenesis.66
Up-to-date research has recently revealed that fucoxanthin, an orange-coloured pigment presented in brown seaweeds, plays a momentous potential in inhibiting the pathogenesis of oxidative stress-induced AMD.67 Pretreatment of fucoxanthin significantly suppresses oxidative stress by reducing ROS and malondialdehyde concentration, nuclear phosphorylated histone deposition and production of senescence-associated β galactosidase.67 Compared with control group, pretreated groups have shown less Aβ deposition, low expression of beta-site APP-cleaving enzyme 1 and the prevention of tight junction disruption.67
These new discoveries could eventually impede Aβ-induced ROS production and oxidative stress and other Aβ-induced injuries in retina cells and also broaden the potential for curing exudative and non-exudative AMD in the future.
Conclusion
Aβ-induced inflammatory activity, ribosomal dysfunction, oxidative phosphorylation dysregulation, spliceosome impairment, angiogenesis and cytoskeleton destabilisation cause numerous damages in the subretinal region, which is associated with the pathogenesis of AMD (figure 1). Future research on the molecular mechanism of Aβ-mediated pathogenesis of AMD may provide novel thoughts about potential therapies of AMD related to Aβ.

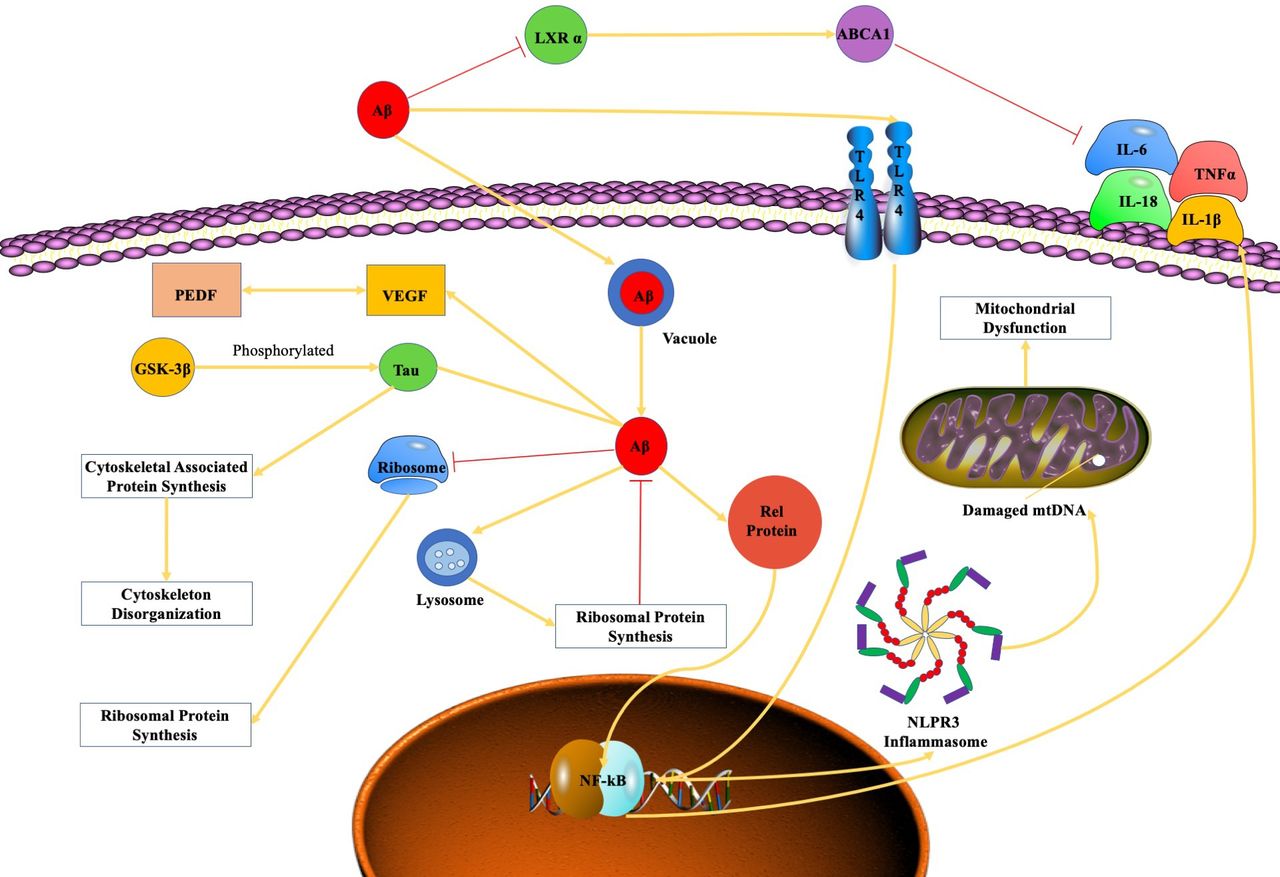{kind=link}
The mechanistic pathwayof amyloid β-peptide in AMD. Aβ, amyloid β-peptide; AMD, age-related macular degeneration; GSK3β, glycogen synthase kinase 3 beta; IL, interleukin; LXR α, liver X receptor α; NF-κB, nuclear factor-κB; PEDF, pigment epithelium derived factor; VEGF, vascular endothelial growth factor.
Ethics statements
References
Footnotes
Contributors MW carried out the searching and collection of publications and drafted the review manuscript. SS participated in collecting the related publications. SJ and XS revised the manuscript. JW, corresponding author, designed and revised the manuscript.
Funding This manuscript was supported by National Natural Science Foundation of China (No. 82070961); Sanming Project of Medicine in Shenzhen (SZSM201812091); International Science and Technology Cooperation Research Project of Shenzhen Science and Technology Innovation Committee (GJHZ20190929145402153).
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.