Article Text

Abstract
Objectives No therapeutic interventions are currently available for autosomal dominant retinitis pigmentosa (adRP). An RPE65 Asp477Gly transition associates with late-onset adRP, reduced RPE65 enzymatic activity being one feature associated with this dominant variant. Our objective: to assess whether in a proof-of-concept study, oral synthetic 9 cis-retinyl acetate therapy improves vision in such advanced disease.
Methods and analysis A phase 1b proof-of-concept clinical trial was conducted involving five patients with advanced disease, aged 41–68 years. Goldmann visual fields (GVF) and visual acuities (VA) were assessed for 6–12 months after 7-day treatment, patients receiving consecutive oral doses (40 mg/m2) of 9-cis-retinyl acetate, a synthetic retinoid replacement.
Results Pathological effects of D477G variant were preliminarily assessed by electroretinography in mice expressing AAV-delivered D477G RPE65, by MTS [3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxyme- thoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium] assays on RPE viability and enzyme activity in cultured cells. In addition to a mild dominant effect reflected in reduced electroretinographics in mice, and reduced cellular function in vitro, D477G exhibited reduced enzymatic RPE65 activity in vitro. In patients, significant improvements were observed in GVF from baseline ranging from 70% to 200% in three of five subjects aged 67–68 years, with largest improvements at 7–10 months. Of two GVF non-responders, one had significant visual acuity improvement (5–15 letters) from baseline after 6 months.
Conclusion Families with D477G variant have been identified in Ireland, the UK, France, the USA and Canada. Effects of single 7-day oral retinoid supplementation lasted at least 6 months, possibly giving visual benefit throughout remaining life in patients with advanced disease, where gene therapy is unlikely to prove beneficial.
- clinical trial
- degeneration
- genetics
- retina
- treatment medical
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Significance of the study
What is already known about this subject?
Oral synthetic retinoid therapy has been shown to be effective in improving vision in autosomal recessive cases of Leber congenital amaurosis (LCA) and retinitis pigmentosa (RP).
What are the new findings?
Oral retinoid therapy can be extended to families so far identified in five countries, in which a unique dominant-acting RPE65 mutation is present causing late-onset RP with choroidal involvement.
How might these results change the focus of research or clinical practice?
Oral synthetic retinoid therapy can be effective in subjects with this late-onset dominant form of disease, where extensive retinal degeneration is evident and where gene therapy would be unlikely to be beneficial.
Introduction
Mutations in genes encoding enzymatic components of the retinoid cycle, reducing the availability of 11-cis-retinal, have been encountered in recessive forms of inherited retinal degenerations (IRDs), retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA).1 In principle, administration of 11-cis-retinal would assist in preventing retinal degeneration in those IRDs where mutations within genes of the retinoid cycle are present. While 11-cis-retinal is unstable and not suitable for pharmacological use, 9-cis-retinal can be used as a replacement, forming iso-rhodopsin which retains function in photon capture and which has been shown, following intravitreal inoculation, to improve vision in dogs with null mutations within the RPE65 gene.2
QLT091001 (9-cis-retinyl acetate) is a late-stage synthetic retinoid replacement therapy being developed for inherited retinal diseases in humans (Retinagenix). It is a prodrug, converted by hydrolysis in vivo to 9-cis-retinol, which is then converted to 9-cis-retinal, thus restoring the key biochemical component of the retinoid cycle. QLT091001 treatment has previously been shown to result in vision improvement (VA) and/or visual field (VF) in subjects with LCA or RP due to recessive mutations in either RPE65 or LRAT genes.3 4 The mutation, D477G in exon 13 of the RPE65 gene is the only mutation within this gene so far identified manifesting autosomal dominant transmission.5 Families displaying this dominant mutation have now been identified in Ireland, the UK, France, the USA and Newfoundland (Professor J. Green, personal communication).5–7
Knock-in mice expressing D477G allele have deficits in retinal structure and function, with disruption of the visual cycle, indicative of decreased RPE65 enzymatic activity. Shin et al reported slower regeneration of 11-cis-retinaldehyde with lower electroretinographic (ERG) a-wave recovery following photobleaching, indicating delayed dark adaptation.8 Choi et al conclude from X-ray crystallographic structural analysis that a possible mechanism for photoreceptor degeneration may involve D477G RPE65 cellular toxicity.9 They also observed retinyl ester accumulation and slower regeneration of 11-cis-retinal after photobleaching in knock-in mice aged 9 months, and slow thinning of the retinal outer nuclear layer accompanied by decline in scotopic and photopic ERG, indicating loss of cone and rod photoreceptor function. Our in vitro data confirm a reduced enzymatic activity of the variant of approximately 25%. However, aberrant splicing of D477G transcripts has recently been suggested as an alternative mechanism contributing to reduced enzymatic activity and ultimately photoreceptor degeneration.10 Hence, in vitro data may not accurately reflect the reduction in enzymatic activity occurring in vivo. Nevertheless, it can be strongly inferred that a component of the disease process in this very slowly progressing dominant retinopathy is limited availability of chromophore 11-cis-retinal and that oral synthetic retinoid therapy may be extendable in improving remaining vision in patients with this genetic subtype of disease, where, as yet, no means of prevention are available for any form of dominant RP.
We report here further observations on the molecular pathology of the disease relevant to a pilot proof-of-concept study in which effects of a single 1 week course of oral retinoid therapy were assessed, and where VFs were significantly improved in three of five patients with advanced disease over periods of 6 months to 1 year. While previous studies on recessive LCA and RP have indicated maximum increases in VA at 2 months following retinoid treatment, responders in the current study were older (average age 62 years) and showed improvements over longer periods lasting over 6–12 months, which we suggest is a possible reflection of the time taken for regeneration of outer segments in remaining photoreceptors in these patients.
Materials and methods
AAV construct design, generation and delivery
Human wild-type (WT) and D477G RPE65 cDNAs with 1400 bp of human RPE65 promoter were cloned into pAAV expression vector (Cell Biolabs). AAV2/9 was generated using a triple transfection system in a stable transfected HEK293 cell line to enable generation of high titre virus (Vector BioLabs); 3 µL of AAV2/9 virus (3.56×1011 vector genomes/mL) was subretinally injected into WT C57BL/6J mice.
Animals
Procedures and animals used in this study were carried out in accordance with regulations set out by The Health Products Regulatory Authority, who are responsible for implementation of EU directive 2010/63/EU. C57BL/6J mice were used in this study. Animals were housed in a specific pathogen-free environment in the University of Dublin, Trinity College.
Murine electroretinographic analysis
Mice were dark-adapted overnight and all manipulations carried out under dim red light as described previously by Palfi et al.11
Transient plasmid transfection
Human WT and D477G RPE65 cDNAs were individually subcloned into mammalian expression vector pRK7 downstream of a CMV promoter. Constructs were purified using QIAGEN Midi Prep kit (Qiagen, France) and transiently transfected into ARPE-19 cells (MTS assay) or HEK293-L cells expressing lecithin retinol acyltransferase (isomerase activity assay) using Lipofectamine 2000 and LTX Reagent, respectively (Invitrogen).
MTS assay
ARPE-19 cells (ATCC #CRL2302) were cultured under standard conditions, plated in 96-well plates at 5×103 cells per well and grown to 50% confluency. Transfection with 40 ng plasmid per well was carried out in reduced serum medium. Transfected cells were incubated for 24 hours and viability assessed by MTS assay using CellTiter 96 AQueous One Solution as outlined by the manufacturer (www.promega.com). This experiment was repeated eight times. For each experiment absorbance readings obtained were standardised to a percentage viability scale by deeming those from cells transfected with WT RPE65 to be 100%.
Isomerase activity assay
HEK293-L cells (3×106) were seeded onto a 25 cm2 culture flask and transfected the following day with 8 µg plasmid using Lipofectamine LTX (Invitrogen).
Forty-eight hours post-transfection, all-trans-retinol (2 µM) was added to the culture and incubated for 24 hours. Cells were harvested, centrifuged and pellets lysed in 200 µL of phosphate buffered saline with 0.2% sodium dodecyl sulfate (SDS), and 300 µL of methanol added. Retinol isomers were extracted with 6 volumes of hexane and analysed by reverse-phase C18 HPLC column and detected by UV absorbance at 325 nm. Peaks were identified based on their characteristic elution times and UV absorbance spectra of retinoid standards. The major substrate of isomerase and the catalytic activity was calculated from the area of the retinyl palmitate and 11-cis-retinol peaks, respectively, using calibration curves determined with standard solutions.
Western blot analysis
Equal amounts of total proteins (20 µg) were resolved by electrophoresis through 10% SDS-PAGE (Mini-Protean TGX precast gels, BIO-RAD) and electrotransferred onto 0.2 µm PVDF (polyvinylidene fluoride) membranes (Trans-Blot Turbo Transfer Pack, BIO-RAD). Membranes were blocked with 5% non-fat dry milk in Tris-buffered saline with 0.1% tween-20 (TBST) and sequentially incubated with a 1:1000 dilution of an anti- RPE65 monoclonal antibody (Millipore, USA) and a 1:5000 anti-β-actin monoclonal antibody (Sigma-Aldrich, USA). After washes with TBST, 1:10 000 dilution of goat anti-mouse IgG alkaline phosphatase conjugate (Sigma-Aldrich) and BCIP/NBT-purple liquid substrate were used to reveal immunoreactivity bands.
Statistical analyses
Data were analysed statistically by one-way analysis of variance or Student’s t-test, as appropriate, using GraphPad Prism 5 software (GraphPad, La Jolla, California, USA) and expressed as mean and SE.
Procedures used for analysis of visual function in proof-of-concept study
Our phase 1b PoC study’s eligibility criteria are noted in www.clinicaltrials.gov. Briefly, males and non-pregnant or non-lactating women from 18 to 70 years of age with better than three Early Treatment Diabetic Retinopathy Study (ETDRS) letters (ie, 20/800) were included. Subjects were treated on an outpatient basis and received the 7-day study treatment in the research clinic under medical supervision.
All subjects underwent visual function testing on both eyes, including best-corrected visual acuity (BCVA measured by ETDRS) and Goldmann visual fields (GVF). Subjects also completed a questionnaire on visual function. Safety assessments included vital signs, pregnancy testing, ECG, clinical laboratory tests (12 hours fasting serum chemistry and haematology, coagulation, thyroid function, serum retinol and urinalysis), biomicroscopy, intraocular pressure, dilated funduscopy; BCVA; adverse events (AEs) and concomitant medications. QLT091001 and metabolite concentrations were also assessed.
For each individual subject, the same test(s) and the same certified VFs examiner were used at screening/baseline and all follow-up visits. Retinal area was calculated from the GVF tests and baseline values for GVF retinal area were the average of all available measurements from the screening/baseline period. Retinal area was interpreted and quantified by a method described by Dagnelie and used in RP clinical research.12 13 The observed retinal area, log transformation of this retinal area and the percentage change from baseline in retinal area for the primary isopter were summarised for all subjects (using eye as an observation) and separately for the right and left eye. One of the five subjects started in Montreal and transferred to Dublin. Only the Dublin data are used for this analysis.
Results
Mouse studies
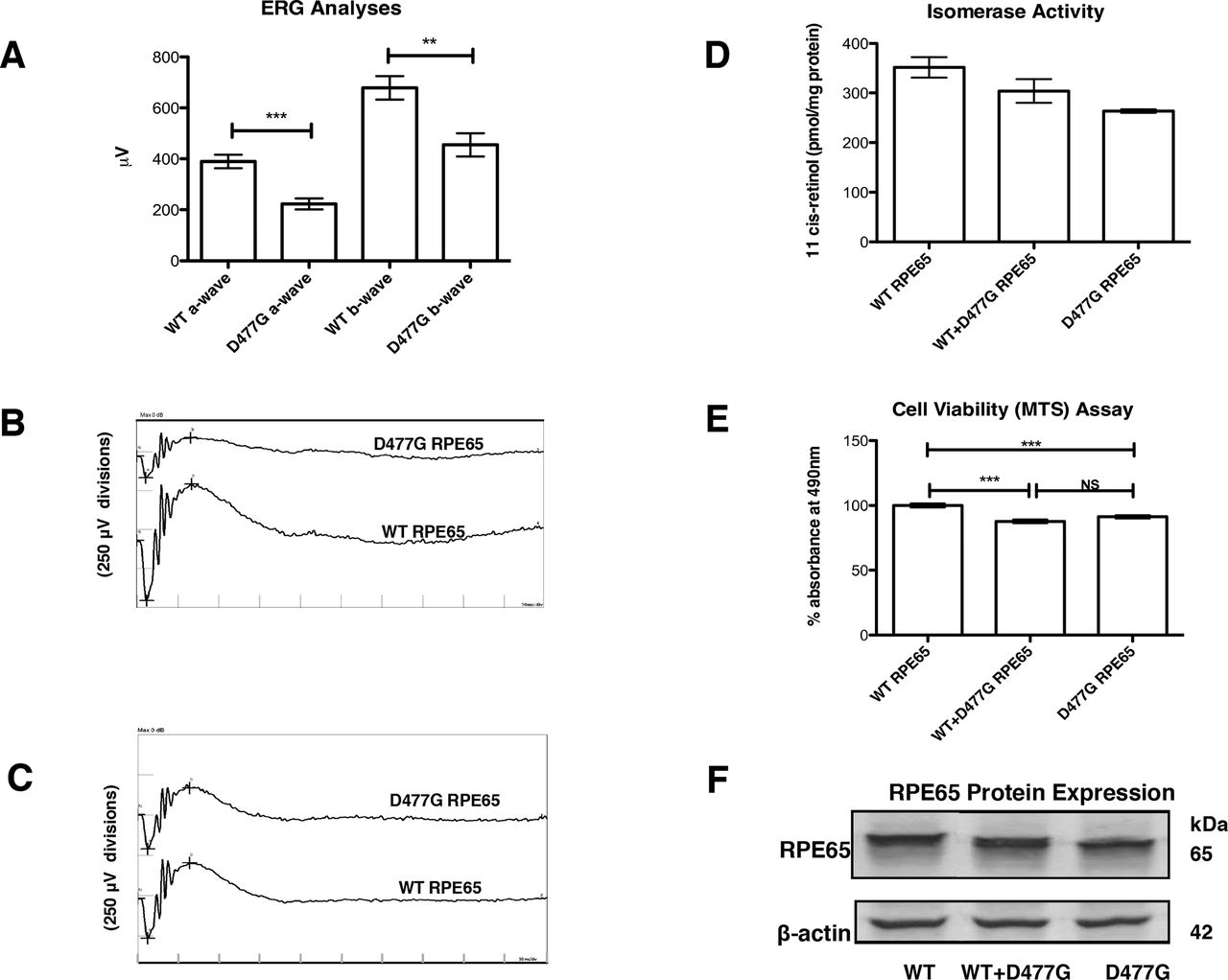
AAV2/9 expressing human RPE65 cDNA or D477G variant driven by the human RPE65 promoter was subretinally injected into WT C57BL/6J mice. Fifty-five days post-inoculation, animals were subjected to ERG analysis (rod, mixed rod and cone and pure cone responses). Amplitude reductions were observed in eyes injected with the D477G variant (n=20, figure 1A–C). Confirmation of presence of normal and mutant transcripts in injected retinas is shown in supplemental data (online supplementary figure S1). Retinal sections from mice injected with AAV expressing the variant protein revealed little or no thinning of the retinal layers in comparison to mice injected with AAV expressing WT protein (not shown), in line with observations on knock-in mice expressing this variant and the lateness of onset and slow progression of this condition in people.5 8 9
Supplemental material
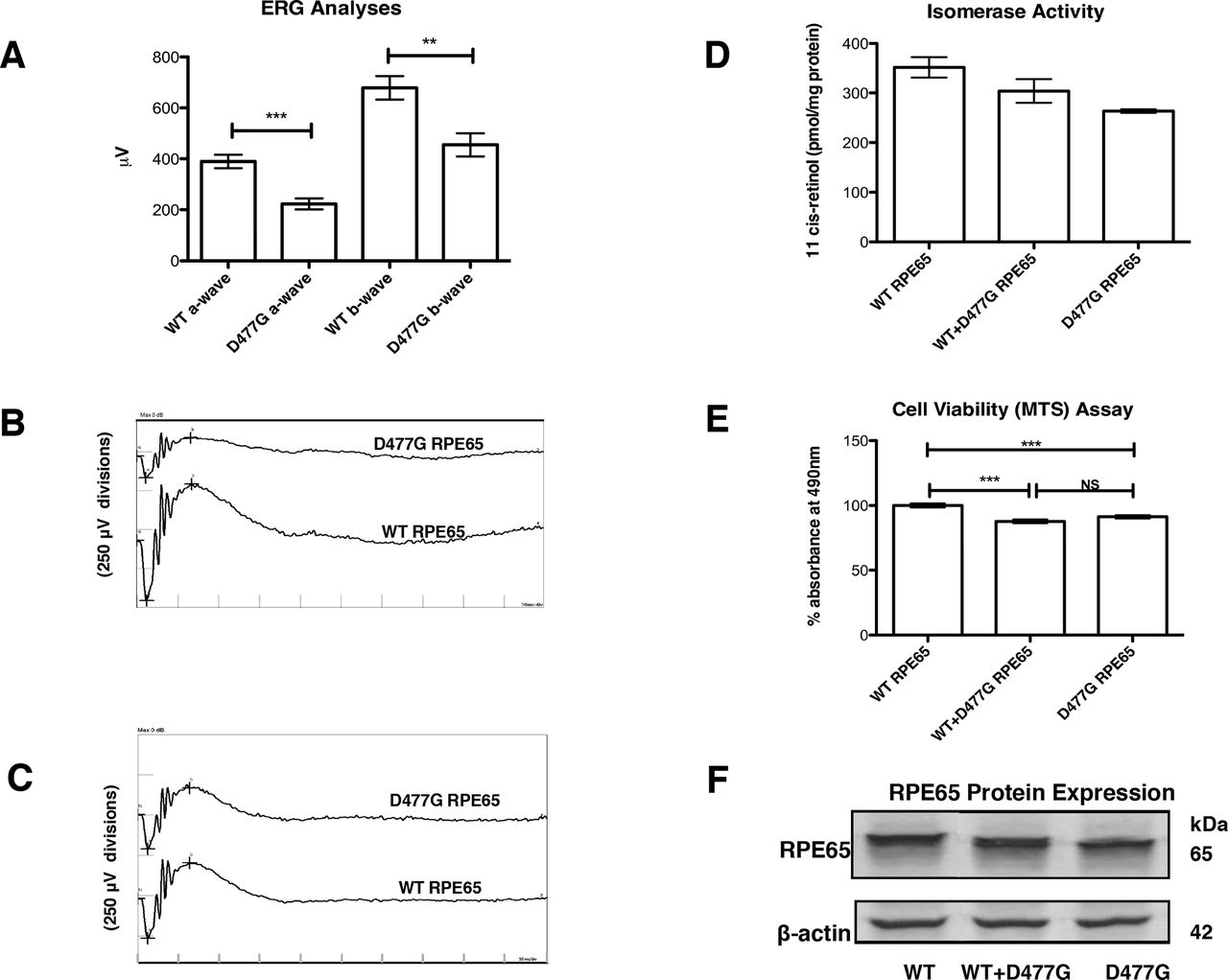
Effect of RPE65 D477G variant on electroretinographic (ERG), RPE viability and isomerase activity in vitro. (A) Composite maximum mixed rod and cone ERG responses from mice (PND 86) following injection of 3 µL virus (3.56×1011 vector genomes/mL) at PND 31 (n=20). (B, C) ERG waveforms of representative mice from (A). (D) Effect of wild-type (WT) RPE65 and D477G RPE65 expression on ARPE-19 cell function as assessed by MTS assay (n=8). (E) Retinoid isomerase activities of WT RPE65 and D477G RPE65 in HEK293-L cells incubated with 2 µM all-trans-retinol (n=2). (F) Immunoblot showing expression levels of WT RPE65 and D447G RPE65 with β-actin as standard.
MTS assays are widely used to quantify cell viability or cytotoxicity in vitro.14 15 Combined data from eight independent assays showed that stressed ARPE-19 cells (see ‘Materials and methods’) transfected with plasmid expressing D477G RPE65 displayed a significantly lower mean absorbance at 490 nm of 91.27% of the value for corresponding cells expressing WT RPE65 (figure 1D). Cells transfected with both WT and D477G RPE65 yielded results which differed significantly from WT RPE65-expressing cells but not from cells expressing D477G RPE65 alone. These data suggest that in this system, mimicking aged RPE, cellular metabolism is compromised by the D477G variant acting in a dominant negative manner over WT RPE65.
We also compared enzymatic (isomerohydrolase) activities of WT RPE65 and D477G RPE65. Equal amounts of plasmids expressing WT RPE65 and D477G RPE65 were transfected separately or combined in a 1:1 ratio into HEK293-L cells and cultured for 48 hours. The enzymatic activities and protein expression levels were verified by isomerase activity assay and western blot analysis, respectively. Levels of retinyl palmitate, the isomerase substrate, were comparable (2.92±0.054 nmol/mg protein) in all transfected cells, showing that adequate isomerase substrate was produced in all transfections. Furthermore, a preliminary HPLC test was performed to ensure that only 11-cis-retinol and not 13-cis retinol or all-trans-retinol would be measured (online supplementary figure S2). D477G RPE65 gave 75.0%±1.5% of the WT RPE65 isomerase activity (263 pmol 11-cis-retinol per mg protein compared with 351 pmol 11-cis-retinol per mg protein) even though protein expression levels were similar (figure 1E,F). A combination of both plasmids showed an intermediate 11-cis-retinol production (86.5%±9.5% of WT, that is, 304 pmol 11-cis-retinol per mg protein), which correlated with an equal expression of each RPE65 protein. We conclude that the D477G mutation decreases the isomerase activity of D477G RPE65 without altering that of the WT RPE65. Thus, in contrast to the effect of D477G variant on cellular metabolism in ARPE-19 cells, we do not observe a dominant negative effect of D477G variant over WT RPE65 enzyme activity. Rather, we suggest, as also proposed by Choi et al, that a facet of D477G other than isomerase function may cause mild cellular toxicity.9
Human studies
We enrolled and treated five participants (aged 41–68 years). No patient was excluded and all enrolled patients completed the study. All patients received seven doses of 40 mg/m2 per day of QLT091001.
Three of the five patients had significant improvements in GVF compared with baseline using log retinal area values (figure 2). Two had bilateral responses, one showing a unilateral response. The magnitude of VF response ranged from a mean increase of 70%–200% in GVF (log retinal area). Maximum responses were at 7 months (two patients) and 10 months (one patient). In the remaining two subjects who were GVF non-responders, one had a significant improvement in VA, from 5 letters at baseline to 15 letters at 6 months.

{kind=link}
{kind=link}
Changes (in %) in Goldmann visual fields (GVF) (log) retinal areas from baseline, after 7 days of oral 9-cis-retinyl ester treatment in dominant RPE65 patients with late-onset retinitis pigmentosa (RP). Patient 10801. This patient aged 68 years shows an improvement in the size of the lll4e GVF area of ~200% and 150% in the R and L eyes after 10 months from baseline, and then a decline to ~45% from baseline at 1 year. The visual acuity (VA) did not change over this time period. The patient noted an ‘improvement in vision’ as a self-reported outcome (SRO). We deemed this patient to be a GVF responder. Patient 10802. This patient aged 68 years shows an improvement in the size of the lll4e area of the L eye only of 90% at 1 month and 70% at 7 months from baseline, and then a decline to baseline at 1 year. The R eye did not respond in this patient. The VA did not change over this time period. The patient noted an ‘improvement in vision’ as an SRO. We categorised this patient as a GVF responder. Patient 10803. This patient aged 67 years shows an improvement in the size of the ll4e GVF area of ~100% and ~70% in the R and L eyes after 7 months from baseline. After 10 months since baseline the ll4e GVF area declines to 40% in both eyes. The VA did not change over this time period. The patient did not experience an improvement on SRO. We concluded this patient is a GVF responder. Patient 10804. This patient aged 65 years shows no improvements in the sizes of the lll4e or the l4e (shown here) GVF areas in the R and L eyes from baseline. However, and importantly, the VA improved from 5 to 15 letters in one eye during the study period from baseline to 6 months (not shown). The patient did not experience an improvement on SRO. We noted that this patient is not a GVF responder but a VA responder. Patient 10805. This patient aged 41 years shows no improvements in the sizes of the lll4e and the l4e (shown here) GVF areas in the R and L eyes from baseline. The VA did not change over this time period. The patient did not experience an improvement on SRO. This patient is an example of a non-responder.
There were no major AEs. With regard to safety and tolerability of retinoid therapy in the current proof-of-concept trial, predominant metabolites following treatment were 9-cis-retinyl palmitate and 13,14-dihydro-9-cis-retinoic acid. Appreciable C4-hour values were also seen for the other retinyl esters and for 9-cis-retinoic acid. The primary metabolite of therapeutic interest, 9-cis-retinol, had intermediate C4-hour values. No deaths, withdrawals or serious or clinically significant AEs occurred during, or following the 7 days of oral treatment. A total of 40 AEs and 20 adverse drug reactions (ADRs) occurred (summarised in online supplementary tables 1-3), all subjects experiencing at least one AE and one ADR. The most common AE was headache, which occurred in all subjects, and all but one event of headache was considered an ADR. Other AEs occurring in more than one subject are included in the supplements.
Supplemental material
Discussion
IRDs remain a major social and economic problem, with many patients suffering from low vision and blindness. IRDs are probably rivalled only by hereditary forms of sensorineural deafness in terms of the genetic heterogeneity they exhibit, with currently ~260+ retinal genes known (RetNet Retinal Information Network: https://sph.uth.edu/retnet/). In addition, up to 30% of IRDs segregate in an autosomal dominant mode, where gene augmentation by gene therapy, feasible in recessive forms of IRD, will generally not be possible. Dominant IRDs will require suppression of dominant-acting alleles at the transcript or gene level rather than gene replacement. These constraints, notwithstanding major recent success in recessive gene therapy for LCA, will render the development of gene-based medicines for most, or all forms of disease both logistically and economically demanding. For the subset of patients who display late-onset autosomal dominant RP with the D477G mutation, periodic oral 9-cis-retinyl ester therapy represents an immediate option. Initial clinical trials of retinoid therapy for autosomal recessive forms of LCA and RP have been encouraging.3 4 In a trial of patients with recessive LCA, 10/14 (71%) showed improvements in GVF retinal area of ≥20%, while 6/14 (43%) showed improvements in VA ≥5 letters or from 0 to seeing any letters on two consecutive visits, in at least one eye.3 In another trial of patients with recessive RP, 8/17 (47%) showed improvements in GVF retinal area of ≥20%, while 11/17 (65%) showed improvements in VA ≥5 letters or from 0 to seeing any letters on two consecutive visits, in at least one eye.4 A number of subjects also provided self-reported outcomes (SROs) and reported being able to walk without their cane, dark adaptation and improved vision in low-light environments. Bittner et al found that intra-visit variabilities of GVF testing in patients with RP are 20% or less, suggesting that values over 20% represent a therapeutic effect.16
Our data indicate here that the synthetic retinoid therapy is extendable to a dominant form of IRD, where patients and families with this variant have so far been identified in five countries. In Ireland, with a population of 4.5 million and where the D447G variant was initially identified, few cases of autosomal recessive RP (or LCA) caused by homozygous mutations in the RPE65 gene have as yet been identified, while over 30 individuals in two families have been identified with the D477G variant. We show significant expansions of the GVF. Three patients increased by 70%–200% compared with baseline, while one of the VF non-responders showed a VA improvement from 5 to 15 letters.
Of great interest is the timing of the improvements. In the LCA study of 16 patients the responding patients improved within 2 weeks and had their maximum increases at 2 months, while in the adult RP study responders improved before 2 months.3 4 In our study reported here, the improvements of the responders were much later, ranging from 6 to 10 months after baseline. Also, our patients were much older, AVG 62 years, compared with 15.7 years in the study by Koenekoop et al, and 28 years in the study by Scholl et al.3 4 It is remarkable that after 62 years of retinal disease, it appears possible to re-activate dormant photoreceptors with a simple oral retinoid. The biology behind this relative delay in our study is not yet known, but we speculate that it takes longer to re-grow an adequate outer segment in our older patients.
One of our laboratory studies suggests a dominant negative effect of the D477G RPE65 allele over the WT allele on RPE cell viability (MTS study), while a second lab investigation shows a decrease in isomerohydrolase activity plus a decrease in 11-cis-retinol levels which can be partially restored by expression of the WT allele. This suggests that there are two fundamentally different kinds of detrimental effects due to D477G allele expression. On the one hand, the mutant protein is mildly toxic to RPE, this toxicity not being alleviated by the presence of WT protein, while on the other hand the D477G mutant enzyme is less efficient than the WT counterpart, but does not interfere with WT enzymatic activity. Further studies will be required to elucidate the toxic nature of the mutant protein. Nevertheless, our study and others indicate alterations of the retinoid cycle and the current investigation showed that an oral retinoid replacement can restore in part this diminished retinoid cycle.8–10
In regard to clinical features of disease caused by the D477G mutation, the retinopathy induced by expression of the D477G variant is of a late-onset retinal degeneration, in some cases not manifesting until the fifth decade of life, and while severely visually handicapped, limited vision remains in some individuals well into the eighth decade.5–7 Given that the beneficial effects of a single 7-day course of synthetic retinoid therapy in the current PoC trial persist for periods of 6–12 months, this form of therapy could provide visual benefit to such individuals throughout remaining life.
In summary, in this study of an autosomal dominant IRD family with an heterozygous RPE65 mutation, we show for the first time a visual rescue following oral 9-cis-retinyl ester administration. A missing retinoid component is replaced, reaching all of both retinas in both eyes. We found delayed yet significant treatment responses in older patients with dominant RPE65 mutations.
Acknowledgments
The authors would like to thank the patients and families for their participation and enthusiasm. We wish to thank Hilary Dempsey and Karen Collins (Research Foundation, Royal Victoria Eye and Ear Hospital, Dublin) for assistance in visual acuity and visual field testing. We also thank Mr. Charles Murray for animal husbandry. The authors deeply regret the passing of collaborator Christian Hamel, during the course of this work.
References
Footnotes
Contributors Contributors PFK: mouse ocular injections, electroretinographic analysis, Clinical Trial Investigator in Dublin, manuscript preparation. MMH: AAV and cDNA vector construction, RNA and PCR analysis, manuscript preparation. A-SK: cell viability assays and manuscript preparation. PB/LG: enzymatic assays and western blot analysis. EO: retinal sectioning. MC: OCT analysis. GJF: experimental design, manuscript preparation. RKK: clinical trial at Montreal, manuscript preparation. PH: experimental design, manuscript preparation.
Funding Supported at Trinity College Dublin by FB-Ireland/Health Research Board (MRCG) and Science Foundation Ireland; at Neurosciences Institute, Montpellier by ANR; RKK financed by FB-Canada, NIH, CIHR. Programme sponsored by Novelion (formerly QLT).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval This two-centre study was carried out at the Montreal Children’s Hospital, McGill University Health Center in Montreal and at the Royal Victoria Eye and Ear Hospital, Dublin, approved by the MUHC and MCH, REB in Montreal and the Ethics and Medical Research Committee of St. Vincent's University Hospital, Dublin.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.