Article Text

Abstract
Objective Systemic activation of the complement system in intermediate age-related macular degeneration (AMD) is understudied. Moreover, links between the presence of reticular pseudodrusen (RPD) and systemic complement dysregulation have not been studied. The aim of this study was to determine if there is a difference in plasma complement factor levels in intermediate AMD compared with controls, and if complement levels are related to the presence of RPD.
Methods and analysis Levels of complement factors C1q (µg/mL), C4 (µg/mL), C2 (µg/mL), Mannose Binding Lectin (ng/mL), C4b (µg/mL), C3 (µg/mL), factor B (µg/mL), factor D (µg/mL), properdin (µg/mL), C3a (ng/mL), iC3b/C3b (ng/mL), Ba (ng/mL), factor H (µg/mL), factor I (µg/mL), C5 (µg/mL), C5a (pg/mL) and SC5b-9 (ng/mL) were measured in plasma.
Results 109 cases and 65 controls were included in the study. Thirty-nine (36%) cases had RPD. Significantly lower systemic levels of: C1q (OR 0.96, 95% CI 0.94 to 0.98), factor B (OR 0.98, 95% CI 0.96 to 0.99), iC3b/C3b (OR 0.97, 95% CI 0.95 to 0.98), factor H (OR 0.99, 95% CI 0.98 to 0.99), factor I (OR 0.83, 95% CI 0.77 to 0.89) and C5 (OR 0.94, 95% CI 0.90 to 0.98) were found in cases versus controls. Significantly elevated levels of: C2 (OR 1.29, 95% CI 1.07 to 1.59), C3a (OR 1.03, 95% CI 1.01 to 1.05) Ba (OR 1.03, 95% CI 1.01 to 1.05) and C5a (OR 1.04, 95% CI 1.02 to 1.07) were found in cases versus controls. Systemic levels of complement factors measured were not related to the presence of RPD.
Conclusions Levels of several systemic complement pathway factors were found to be altered in intermediate AMD. Systemic levels of complement factors were not related to RPD.
- epidemiology
- inflammation
- macula
- retina
- immunology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Alterations in systemic levels of complement factors have been previously described in patients with age-related macular degeneration (AMD). However, prior studies have not focused specifically on intermediate AMD. Moreover, links between the presence of reticular pseudodrusen (RPD) and systemic levels of complement factors have not been studied.
What are the new findings?
Altered systemic plasma levels of several complement factors from the alternative and the classical complement pathway were found in patients with intermediate AMD compared with controls. Complement factor levels were not related to the presence of RPD.
How might these results change the focus of research or clinical practice?
There is a need for more studies of proinflammatory factors in patients with the early and intermediate forms of AMD to determine markers that may predicts progression to either the neovascular or geographical atrophy forms of advanced AMD.
Introduction
Age-related macular degeneration (AMD), a progressive degenerative disease of the macula, is a leading cause of vision loss in individuals over 60 years. It is estimated that by 2020, 196 million individuals worldwide will be affected by AMD.1 The hallmark of AMD is the deposition of extracellular material called drusen between the basal lamina of the retinal pigment epithelium (RPE) and Bruch’s membrane.2 In the last three decades, other deposits called reticular pseudodrusen (RPD) have been described in patients with AMD.3–6 Unlike drusen, RPD, which are described as yellowish, interlacing macular lesions on colour fundus photography, are located between the RPE and the photoreceptors.7 8 These deposits are often referred to as subretinal drusenoid deposits.9 10
Advances in imaging modalities described in detail elsewhere8 11 have facilitated improved detection of RPD and prompted more research into the clinical significance of these subretinal lesions.12 13 The presence of RPD is now recognised as a distinct AMD phenotype.14 Several investigators have reported that the presence of RPD is clinically and pathophysiologically related to the advanced forms of AMD.15 16 Moreover, a relationship between RPD with several risk factors, including increasing age, female sex and genetic variants, has also been described.8 17–22 Investigators have suggested that the pathophysiology of RPD formation is at the level of the RPE.7 23 Pathological changes in the choroid24–29 and dysregulation of cholesterol homeostasis7 have also been shown to be related to RPD.
The complement system has a pivotal role in AMD.30 31 As described by Holers,32 the complement system has three main functions: it defends the host against infections, it bridges innate and adaptive immunity, and facilitates disposal of immune complexes, apoptotic cells and the products of inflammatory or traumatic injury. The effector functions of the complement system are initiated by the classical (CP), lectin (LP) and alternative (AP) pathways, and the effects of activation of any of the three are greatly increased by engagement of the amplification loop (figure 1). The three pathways converge at the point of C3 activation through the generation of C3 convertases. These are activating enzymes that cleave C3 to C3a and iC3b/C3b. iC3b/C3b binds to the C3 convertase to form a C5 convertase. Cleavage of C5 results in the formation of C5a and C5b. C5b is a component of the terminal complement complex (C5b-9), also designated the membrane attack complex (MAC), a multimer which can lyse cells and can activate signalling pathways in nucleated cells.33 C3a and C5a mediate inflammation and chemotaxis. While the CP and LP are engaged when ligand recognition molecules bind to their targets and engage coassociated serine proteases, the AP is constantly being slowly activated in plasma and on biological surfaces through a process designated tickover.34 To prevent uncontrolled complement activation and damage to self-cell surfaces, soluble and cell-associated complement proteins provide downregulation of the complement system at several levels during the activation process.
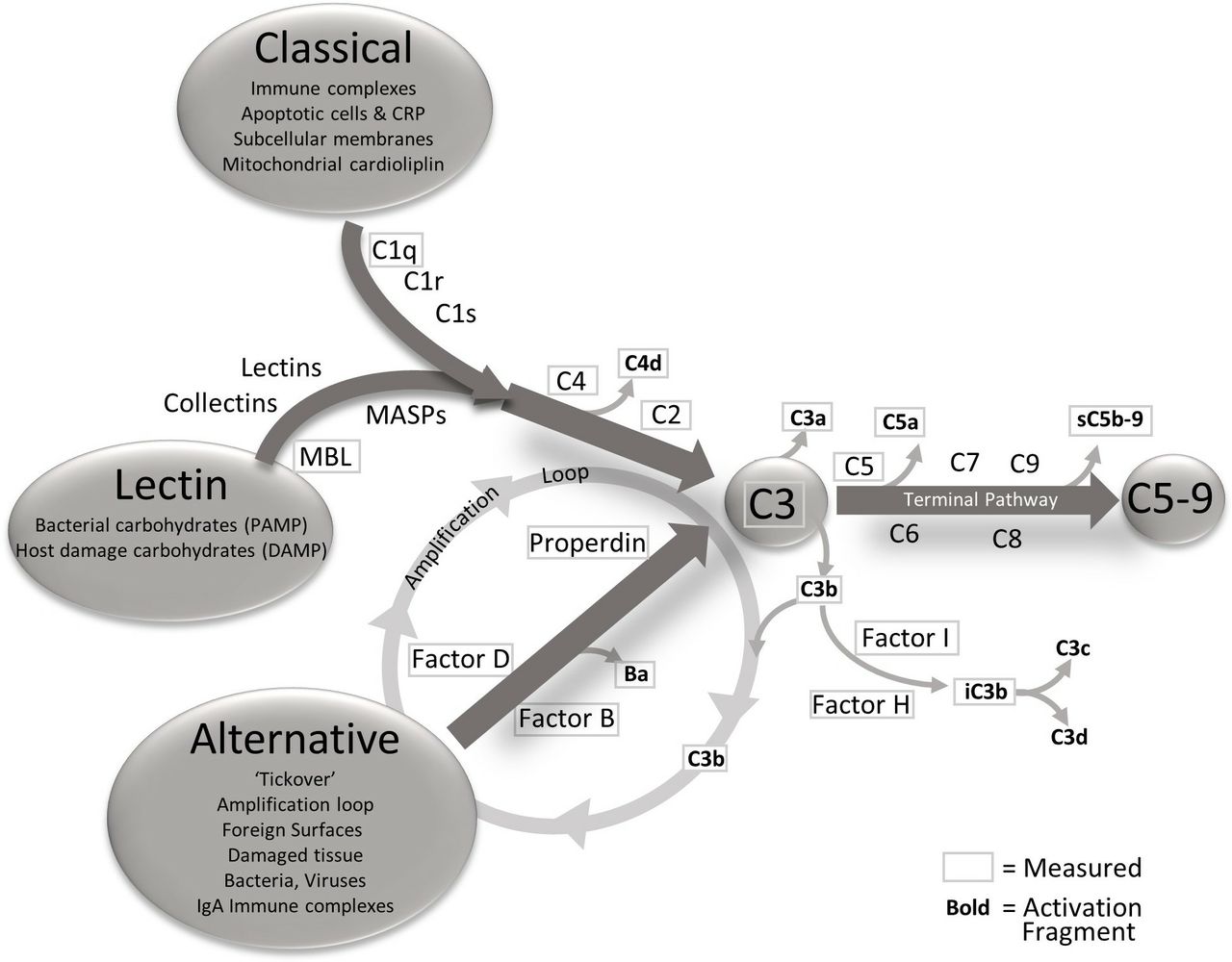
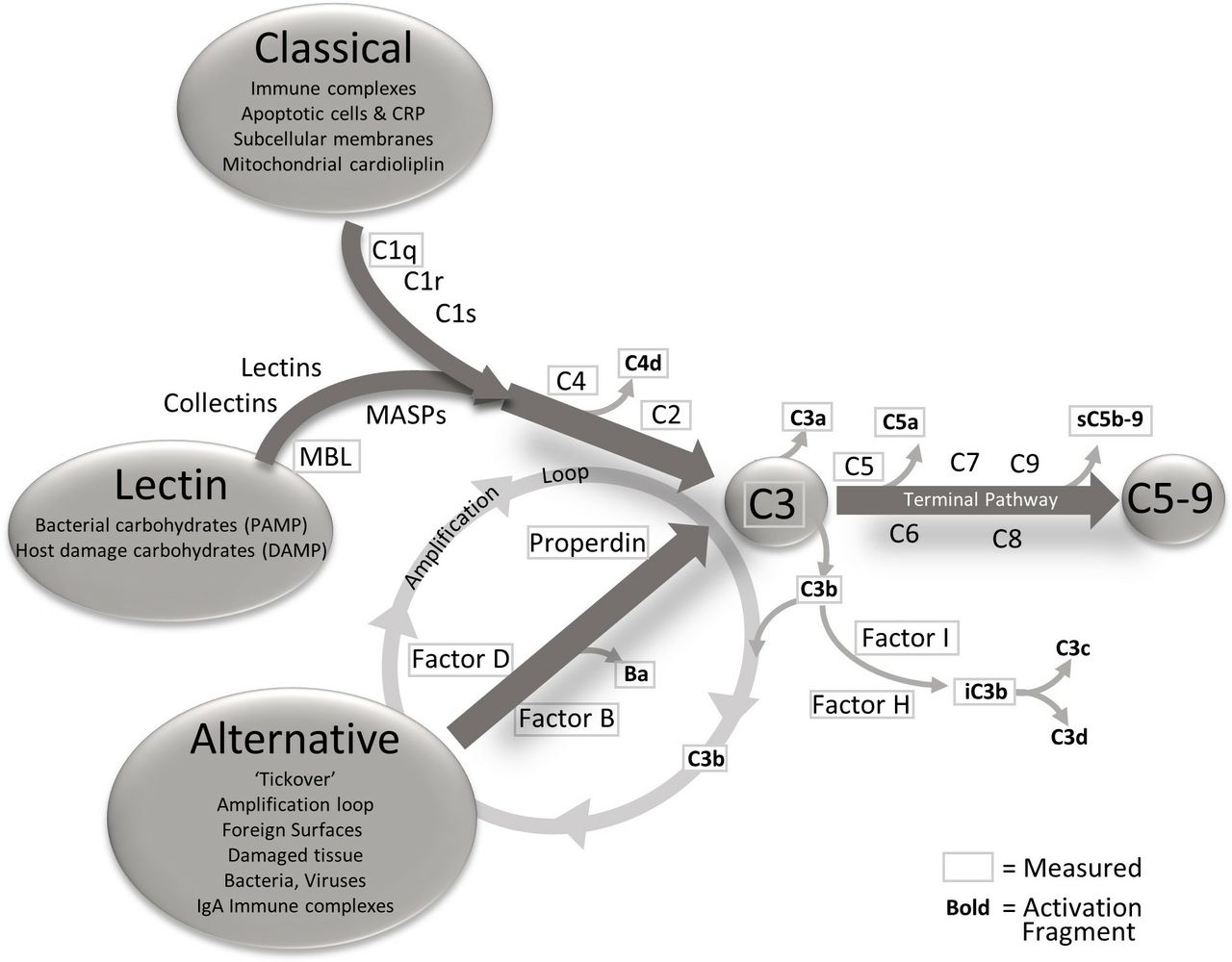{kind=link}
The complement pathways, triggers for activation and analytes measured. The complement system has three activation pathways, classical, lectin and alternative. These three pathways are activated differently but all connect at the central point C3. The three pathways are interrelated by the action of alternative pathway as an amplification loop for the classical and lectin pathways. After C3, the complement system enters the terminal pathway, culminating in the membrane attached complex (also known as C5b-9). As activation flows through the cascade, the components are cleaved yielding activation fragments that are released into circulation. The components and activation fragments measured in this study are designated with a frame and encompass the whole cascade. CRP, C reactive protein; DAMP, damage-associated molecular pattern; PAMP, pathogen-associated molecular pattern; MBL, Mannose Binding Lectin.
Our group35 and others36–44 have described alterations in systemic levels of complement factors in patients with AMD. However, most of these existing studies have not focused specifically on intermediate AMD. Moreover, links between RPD and systemic dysregulation of the complement system have not been studied. Our research is focused on a carefully characterised group of patients with the intermediate form of AMD. We addressed two research questions in our study: (1) to determine if there is a difference in systemic levels of a panel of activation pathway, regulatory and effector complement factors in patients with intermediate AMD compared with controls without AMD and (2) to determine among patients with intermediate AMD if alterations in systemic complement factor levels distinguish patients with and without RPD.
Methods
Overview of the Colorado AMD registry
We conducted this case–control study by use of the records and samples from an AMD research registry and repository developed by the Department of Ophthalmology at the University of Colorado School of Medicine. Patients with AMD and controls with recent cataract surgery but no AMD who receive care at the UCHealth Sue Anschutz-Rodgers Eye Center are invited to be part of the registry. Informed consent is obtained from the subjects after explanation of the nature and possible consequences of the study. Enrolment into the registry is ongoing.
Patient and public involvement
We encourage patients with AMD to be part of our registry. We update our patients on our research achievements in a brochure which is made available in our clinics. We also include information related to our registry in a newsletter produced by UCHealth.
Recruitment, exclusion/inclusion criteria
The recruitment, exclusion/inclusion criteria and informed consent of each participant are described in detail elsewhere.35 45 46 In summary, each case and control is consented for: (1) review of the medical history, (2) collection of an ethylenediaminetetraacetic acid (EDTA) plasma sample (collected from controls at least 1 month after cataract surgery) and (3) review and disease phenotype classification of image data to include a colour fundus photo, fundus autofluorescence (FAF), near-infrared fundus reflectance (NIR) and spectral domain optical coherence tomography (SD-OCT). Ocular exclusion criteria for cases and controls are: panretinal photocoagulation or antivascular endothelial growth factor injections for diabetic retinopathy, branch and central retinal vein occlusion, any active ocular inflammatory disease or a severe decrease in visual acuity secondary to a preexisting severe retinal disease other than AMD.
Image review
The images, described above, on cases and controls, performed on enrolment into the study are reviewed by two vitreoretinal specialists (MTM and FSS) at a twice monthly research image review session conducted in our department. Classification is assigned in the absence of knowledge of the complement values. The image review is focused on an examination of the anatomic macula, which includes the entire area between the retinal vascular arcades. The images are categorised into early, intermediate or advanced AMD using the classification described by Ferris et al.2 The absence of drusen is confirmed in the cataract controls using similar imaging techniques. Discrepancies are resolved by a third vitreoretinal specialist (NM). The presence or absence of RPD is also determined. We followed guidelines for the definition of RPD described by other authors.18 27 47–50 On CFP, RPD are defined as confluent deep retinal lesions forming an interlacing ribbon-like network.51 On NIR, RPD are considered as groups of hyporeflectant lesions against a mildly hyper-reflectant background.18 50 On FAF, RPD are defined as a group of ill-defined, hypofluorescent lesions against a background of mildly elevated autofluorescence.49 On SD-OCT, RPD are identified as an accumulation of hyper-reflective material in the outer retinal layer anterior to the RPE.48 Our definition of the presence of RPD is RPD observed on FAF and/or NIR imaging and confirmed on SD-OCT.
Study-specific inclusion and exclusion criteria
To address the research questions, a second image review was conducted by NM and AGP on the records of non-Hispanic white AMD cases (n=118) with image confirmed intermediate AMD (advanced and early cases were removed from the analytic dataset) and 65 controls with image confirmed no AMD recruited into the registry between July 2014 and December 2017. This second review was conducted to specifically: (1) confirm no misclassification of the presence or absence of RPD and (2) to apply exclusions specific to this study, other retina comorbidities (n=5) and unilateral RPD (n=4). Following this review, 109 cases with intermediate AMD and 65 controls remained in the final analytical dataset. Risk factors in this dataset included: age, sex, family history of AMD (defined as a parent or sibling diagnosed with AMD per patient report), body mass index and select comorbidities.
Collection and processing of the plasma sample
Following phlebotomy, the EDTA tube was spun at 3000 revolutions per minute (rpm) in a cooled (4°C) centrifuge for 10 min to isolate plasma. The average time from phlebotomy to spin was low at 2.6 min±1.7 SD, range 0 to 10 min- an approach which minimised ex vivo complement activation. The time to spin was not different between cases and controls. The aliquots of plasma were immediately stored in a −80°C freezer.
Complement factor panel analysis
The frozen cryovials were transferred to Exsera BioLabs, a CAP/CLIA certified laboratory for analysis, housed on the University of Colorado Anschutz Medical Campus in the Division of Rheumatology. Plasma Complement factors levels C1q, C4, C2, Mannose Binding Lectin (MBL), C4b, C3, factor B, factor D, properdin, C3a, iC3b/C3b, Ba, factor H, factor I, C5, C5a and SC5b-9 were measured. The C5b-9, also known as the MAC, has the potential to lead to the lysis of susceptible membranes. The complement inhibitor vitronectin (aka S-Protein) controls the process by forming the SC5b-9 (soluble C5b-9) complex, maintaining it in circulation and inhibiting its lytic abilities. Measurement of complement components and activation fragment levels was performed by two methods. The Ba, C3a and SC5b-9 levels were measured by ELISA (Quidel, San Diego, California, USA). The remaining measurements were performed by multiplex Luminex immunoassays (MilliporeSigma, Burlington, Massachusetts, USA). For both methods, the assays were run masked to case–control status. The methods were optimised and validated to the level required for regulated laboratory analysis.
All analyses were performed in duplicate with the resulting mean values reported. For the multiplex Luminex data, the mean fluorescent intensity was the raw value, and for the ELISA analysis the raw value was optical density. Standard curves and a four parameter parametric curve fit were used to calculate the absolute quality in ng/mL or µg/mL, as appropriate. Three quality controls (QCs) were included in each run, including at least one laboratory developed and characterised QC. The QCs were monitored for performance and for all testing in the study the values returned were within required parameter, demonstrating assay performance. Human plasma reference ranges for the analytes tested have been determined within Exsera by the measurement of greater than 100 normal that was inclusive of gender and race. During the validation, the interassay and interassay precision were tested for all the analytes at three concentrations across the analytical measurement range over 18 replicates. The result were as follow: Luminex panel 1 (C2, C4b, C5, C5a, factor D, MBL and factor I); intra-assay max 12.2% CV, average 6.8% CV and for interassay max 15.2% CV, average 8.8% CV : Luminex panel 2, (C1q, C3, C3b/iC3b, C5, factor B, factor H and properdin) intra-assay max 10.9% CV, average 4.7% CV; interassay max 19.6% CV, average 12.5% CV: ELISAs intra-assay max 10.5% CV, average 5.3% CV; interassay max 16.7% CV, average 9.8% CV.
Statistical analysis
Descriptive statistics included percentages, means and SD. We examined the relationship of categorical risk factors between cases and controls using the χ2 test or Fisher’s exact test where indicated. Differences in continuous variables were compared using two-sample t-tests and univariate logistic regression. The OR was used as a measure of association and is presented with 95% CIs. We adjusted for multiple comparisons testing using the false discovery rate and for potential confounding using multivariable logistic regression analysis. All analyses were performed using SAS software (V.9.4, SAS). A p<0.05 was considered statistically significant throughout the analysis.
Results
We show in table 1 the differences in demographic variables and select comorbidities between subjects with intermediate AMD (n=109) and controls (n=65). Significantly higher frequencies of a family history of AMD and a history of asthma were found in cases with intermediate AMD compared with controls. The median age was also significantly higher in the intermediate AMD group compared with controls.
Differences in clinical characteristics between subjects with intermediate AMD and controls
In table 2, we demonstrate differences in systemic complement factors between subjects with intermediate AMD compared with controls. The multivariable logistic regression adjusted for potential confounding and multiple comparison testing results are also presented. As shown, following these adjustments, levels of C1q and C2, complement components of the CP were significantly different between cases and controls. We also found alterations in complement factors linked with the AP, specifically lower levels of Factor B, higher levels of C3a and Ba, and lower levels of iC3b/C3b. Levels of the complement inhibitory proteins were also altered, with lower levels of factor H and factor I observed in cases compared with controls. Significant differences in the terminal complement pathway complement component protein, C5 and the activation fragment, C5a were also found in subjects with intermediate AMD compared with controls.
Complement levels for cases with intermediate AMD versus controls
In table 3, we also present differences between demographic and clinical characteristics among subjects with intermediate AMD with and without RPD. As shown, 39 of 109 (36%) cases of intermediate AMD had evidence of RPD. There was no significant difference in age between subjects with and without RPD. The proportion of subjects with a family history of AMD was significantly higher in subjects with RPD compared with subjects without RPD. The presence of RPD was also significantly associated with a history of treated hypertension.
Differences in characteristics of those with and without RPD in the Intermediate AMD group
In table 4, differences in systemic complement levels in intermediate AMD subjects with and without RPD are shown. None of the systemic complement factors were significantly different between cases with RPD and cases without RPD.
Complement levels in subjects with intermediate AMD stratified by the presence or absence of RPD
Discussion
There were two key findings from this study. First, we found significant alterations in systemic levels of complement factors (as measured by 17 complement factors) related to AP and CP and terminal complement pathways in cases of intermediate AMD compared with controls. Second, cases with and without RPD had similar levels of systemic complement factors.
In agreement with results from other investigators,36–44 our study suggests that systemic complement factor levels differ in patients with AMD compared with controls without AMD. Most of the previously described studies of AMD-related systemic complement activation have focused on individuals who had the advanced stages of AMD or AMD categories were examined as a composite outcome. One other group of investigators studied a similar early-stage AMD group and found in keeping with the results of our study that C3a was elevated in cases compared with controls.38 Our study is distinguished by the fact that we were focused in our methods on precise phenotyping of our study group.52 Moreover, we measured a large panel of complement factors reflecting activation of all complement pathways.
We suggest a novel role for the CP in intermediate AMD. Low levels of C1q (indicating consumption of the protein), a macromolecular complex (C1q, C1r and C1s) and the first protein of the complement cascade (figure 1), were observed in the intermediate AMD group compared with controls. This complement factor is linked with the clearance of immune complexes, as well as damaged and apoptotic cells. C reactive protein, an acute phase reactant, can also bind and activate C1q.53 Our results highlight a potential role for the CP in intermediate AMD.
Aligned with the existing literature, we found evidence of a role for the AP in AMD, reviewed in Lambert et al54; specifically, we observed lower levels of factor B, indicating cleavage by factor D and higher levels of the complement activation fragments, C3a and Ba, between cases and controls. In agreement with results from Reynolds et al,36 who studied advanced AMD, we found factor H, one of the complement inhibitory proteins, to be lower in cases compared with controls (table 2). We also found lower levels of another complement regulator, factor I, to be linked with intermediate AMD. Significant markers of terminal complement pathway activation observed were lower levels of C5, indicating cleavage and higher levels of C5a, an anaphylatoxin and chemotactic factor.
We found that none of the complement factors measured in our study distinguished subjects with intermediate AMD who did and did not have RPD. The role of the complement system in intermediate AMD is understudied. It is noteworthy that Wu et al21 studied an Australian cohort of 300 subjects with intermediate AMD defined also by the Ferris et al classification.2 This study, from 2016, evaluated RPD in this select phenotype of AMD. It is of interest that the single-nucleotide polymorphisms of rs1061170 (Y402H) in the complement factor H (CFH) gene—a marker of dysregulation of the AP was not associated with RPD.
Other investigators have described a role for local dysregulation of the complement system in AMD. Indeed, Mullins et al found MAC levels to be higher in the choroid in CFH high-risk genotypes from human donors.55 It is uncertain whether local, systemic or a combination of local and systemic dysregulation of the complement system is driving the pathology of AMD, reviewed in Warwick et al.31 The advantage of studying systemic alterations in complement levels is that the samples are easily obtained, and levels can be studied over time whereas local ocular complement activation cannot be studied in vivo. The evidence presented here suggests that the local ocular changes seen in intermediate AMD occur in the setting of a systemic inflammatory milieu in which systemic dysregulation of the complement system plays an important role. Our findings highlight the importance of further research to understand how aberrant complement activation affects the natural history of AMD. As reviewed by Tomlinson and Thurman56 and Trouw et al,57 the complement system has been shown to have a role in in other comorbidities. Indeed, therapeutic complement inhibitors have been introduced as treatments in several of these systemic diseases.56 57
Strengths of our study include the careful phenotyping of the intermediate AMD cases and controls using multimodal imaging, the meticulous collection of the plasma samples58 and short time to freezer storage. We also acknowledge that there are limitations to our study. One key set of risk factors not examined were genetic polymorphisms of the complement pathways—well recognised as significant risk factors for AMD.54 As reported by many authors,43 genetic polymorphisms of the CFH and ARMS2 genes contribute significantly to the development of AMD. Indeed, CFH is reported as being one of the strongest genetic risk factors for the development of AMD, accounting for 50% of the attributable risk for the disease.54 It is suggested that polymorphisms of the CFH gene attenuate the inhibitory function of factor H (figure 1). Indeed, several investigators have found a relationship between polymorphisms in the AMD susceptibility genes with systemic complement activation.41 42 44 We also acknowledge that we did not match on age in our study design. However, we adjusted for age in the multivariable logistic regression analysis to account for the difference between cases and controls. As we continue to develop our cohort of individuals with the early stages of AMD, we will also investigate genetic polymorphisms of the complement pathways and the relationships of these variants with systemic complement levels. We will also account for the important contribution of an individual’s complotype to AMD.59
In summary, we suggest a significant association of systemic dysregulation of the complement system with intermediate AMD. Our study suggests a contribution of not only the AP and terminal complement pathways but also a novel role for the CP in intermediate AMD. Another novel finding was that there was no association between systemic complement levels and AMD cases with and without RPD. There is a need for more investigation of patients with the early and intermediate forms of AMD so that treatments can be introduced before the individual progresses to advanced AMD. Although several clinical trials are ongoing, finding an intervention that attenuates the effects of complement activation in AMD has thus far been elusive.30 60
Future research should also focus on the study of individuals who present early in the course of AMD with panels of complement/inflammatory factors collected longitudinally to identify if there is a complement/inflammatory profile linked with progression to either the neovascular or geographical atrophy stages of AMD. Ultimately, by combining genetic, environmental, demographic, behavioural and systemic biomarker profiles, it may be possible to develop a personalised approach to the treatment of individuals early in the natural history of AMD.
References
Footnotes
Contributors AML, NM and AGP planned and designed the study. AML was responsible for conduct of the study and collection of data. AML and NM were responsible for the overall coordination of the study. BDW and JLP conducted the data analysis. AAF-A measured the complement factors. MTM, FSS, NM and AGP reviewed the images. VMH and AAF-A provided consultation on the interpretation of the complement data. All authors contributed to the interpretation of the data, drafts of the manuscript and editing of the final paper.
Funding Support from a Challenge Grant to the Department of Ophthalmology from Research to Prevent Blindness and the Frederic C. Hamilton Macular Degeneration Center.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The registry is approved by the Colorado Multiple Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon request.