Article Text

Abstract
Objective To explore the impact of weekly transcorneal electrical stimulation (TES) over a 6-month period as a treatment for retinitis pigmentosa (RP).
Methods and analysis A prospective open-label observational trial was carried out assessing weekly TES in participants with RP for a period of 6 months followed by observation for a further 6 months. Clinical examination and investigations were carried out at 3 monthly intervals for a total of 12 months. The primary outcome measure explored safety through a descriptive analysis of adverse effects with secondary outcome measures evaluating structural and functional efficacy.
Results Seven male and seven female participants with RP aged 18–80 years were recruited. TES was well tolerated with no serious adverse events reported. Two participants reported transient foreign body sensation and one participant had discomfort underneath the skin electrode. Following 6 months of TES, best-corrected visual acuity increased by 1.1±1.4 letters in the control arm and 0.93±1.4 letters in the treated arm. Central microperimetry threshold sensitivity rose by 0.02±0.5 decibels (dB) and 0.37±0.4 dB and Goldmann visual field volume by 0.16±0.09 steradians (sr) vs 0.22±0.12 sr for the control and treated eye, respectively. There was no statistical significance seen between eyes following the treatment or observation period.
Conclusion This small open-label clinical trial showed that TES was safe and well tolerated in patients with RP. Visual function measurements at 6 months demonstrated no significant difference between the control and treated eyes. The results justify a larger clinical trial over a longer period of time in order to identify any treatment effect.
- treatment other
- retina
- genetics
- degeneration
- clinical trial
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Transcorneal electrical stimulation (TES) induces photoreceptor preservation in murine models of retinal degeneration. A pilot study in eight human participants undergoing weekly TES for 6 weeks showed significant improvements in their kinetic perimetry and scotopic electroretinography.
What are the new findings?
This study provides further evidence of the safety profile of TES with no serious adverse effects noted. The most common side effect was transient self-resolving foreign body sensation. Psychophysics and structural measures of optical coherence tomography and fundus autofluorescence did not reveal any significant change when comparing the control with the treated eye.
How might these results change the focus of research or clinical practice?
The growing evidence supporting the safety of TES justifies a larger clinical trial of longer duration. Participants should ideally show demonstrable deterioration in the control eye to identify any potential treatment effect.
Introduction
Retinitis pigmentosa (RP) describes a group of genetically heterogeneous retinal dystrophies, typically characterised by a primary loss of rods with secondary cone involvement. There remains no established therapy for RP and thus the condition frequently leads to substantial visual impairment in the majority of patients. One proposed strategy to halt or slow progressive photoreceptor loss is the administration of transcorneal electrical stimulation (TES). TES confers preservation of outer nuclear layer thickness and electroretinography function (ERG) in murine models of retinal degeneration after 6 weeks of therapy.1 Similarly, retinal ganglion cell death following optic nerve transection can be significantly reduced following a single session of TES.2 These benefits are believed to result from the upregulation of specific neurotrophic factors.3 4 Several groups have demonstrated an increase in ciliary neurotrophic factor, fibroblast growth factor alpha and brain-derived neurotrophic factor following electrical stimulation.5–7 Interestingly, participants receiving electronic subretinal implants have shown visual improvement in areas distant from the implant further suggesting that electrical stimulation may induce benefit through an amplification of neurotrophins.8 9 Further contribution may come from improved chorioretinal circulation, which has been shown to increase following TES in normal participants,10 although it is unclear whether this is in addition or secondary to neurotrophic upregulation.
TES has been reported to improve visual function in humans with retinal artery occlusion, non-arteritic ischaemic optic neuropathy and traumatic optic neuropathy,11–13 as well as inherited retinal dystrophies, such as Best vitelliform macular dystrophy.14 In RP, an initial pilot study assessing six sessions of weekly TES in eight participants showed significant improvements in kinetic perimetry and scotopic ERG function.15 However, a subsequent follow-up study did not reproduce psychophysical improvements but revealed gains in the photopic b-wave amplitude.16 Given its low-risk profile and ease of application, TES remains an attractive potential therapeutic option. Here, we report the results of the Transcorneal Electrical Stimulation for the treatment of retinitis pigmentosa Open Label multicentre UK study exploring primarily the safety and secondarily the efficacy of TES in participants with RP recruited for a 12-month period commencing with 6 months of weekly TES.
Subjects and methods
Fourteen participants with rod-cone dystrophy were recruited into an open-label interventional trial from the retinal genetics clinics at the Oxford Eye Hospital and Moorfields Eye Hospital. Clinical diagnosis of RP, defined in this study as an inherited retinal disease of rod-cone dystrophy, was through history, posterior segment examination, fundus autofluorescence (AF), optical coherence tomography and, in most cases, full-field electroretinography. Seven participants had recognised mutational variants on next-generation sequencing, as illustrated in table 1. In addition to a diagnosis of RP, inclusion criteria were 18–80 years of age, a visual acuity >0.02 (Snellen decimal) and sufficient motor skills to apply the device independently. Exclusion criteria were the presence of diabetic retinopathy, previous retinal vascular occlusion, glaucoma, previous retinal detachment, previous silicone oil tamponade, macular oedema, corneal disease, neovascularisation of any origin and female participants who were pregnant or planning pregnancy during the course of the study. In addition, simultaneous participation in another interventional study, and some forms of mental illness, which might interfere with study participation, were exclusion criteria.
Demographics of all participants
Following recruitment, participants were assessed at a baseline visit and underwent phosphene threshold determination and a first session of TES. Follow-up assessments were three monthly through one total year of participation. TES was administered weekly under the supervision of a trained member of the research team for 30 min for the first 6 months over a minimum of 24 sessions of TES. A discussion regarding any adverse effects experienced took place each week prior to TES. Participants were then observed for a further 6 months without any TES at 3 monthly intervals.
Data were recorded in the clinical notes and the Case Report Form. Data monitoring was provided by an independent Contract Research Organisation, STZ Eyetrial, based at the University Hospital Tuebingen and was conducted within the stipulations of the International Conference on Harmonisation (Geneva, Switzerland). The study was conducted according to the tenets of the Declaration of Helsinki (2008) and Good Clinical Practice. All participants gave informed consent prior to enrolment. Public trial registration was through clinicaltrials.gov (NCT01847365).
Visual function was measured at 3 monthly intervals using ETDRS best-corrected visual acuity (BCVA) at 4 m, Goldmann visual field analysis and MAIA microperimetry (MP) (Centervue, Padova, Italy). MP was performed prior to dilation, following 20 min of dark adaptation. The MAIA combines a scanning laser ophthalmoscope with perimetry—a Goldmann size III (0.7 degree diameter) target was projected onto the ocular fundus via an artificial entrance pupil measuring 2 mm as previously described.17 Average threshold sensitivities were determined for the central 10 degrees (10–2 test) of the field of vision. Test accuracy was ≥85% for each test.
Retinal structure was assessed using Heidelberg Spectralis spectral domain optical coherence tomography (OCT) in accordance with the manufacturer’s protocols. Fundus AF imaging was performed with a Heidelberg Spectralis OCT-confocal scanning laser ophthalmoscopy machine with the BluePeak blue laser AF module (Heidelberg Engineering GmbH, Heidelberg, Germany).
Electrical stimulation
TES was administered using sterile single-use DTL electrodes (OkuEl) positioned onto a rigid semicircular frame mounted onto the OkuSpex, a metallic spectacle-like frame (see online supplementary figure 1). The electrode was placed into the inferior fornix. The participant’s ipsilateral forehead was cleaned using 70% isopropyl alcohol and electrode contact paste with a skin counter electrode placed over the area. Electrodes were attached to the OkuStim, a neurostimulation device manufactured by Okuvision GmbH. Individual phosphene threshold was established using the OkuStim software (V.1.4.4.0). Stimulation parameters included pulses of 5 ms positive deflection and 5 ms negative deflection with a frequency of 20 Hz. The current delivered was slowly titrated up from 0.02 mA, in increments of 0.01–0.05 mA to a maximum current of 1.0 mA, until participants perceived phosphenes. This phosphene threshold was then rechecked a further two times using a staircase method. The average of the three values formed the participant’s individual phosphene threshold (IPT). The IPT was recorded onto the participant’s unique USB stick, which was attached to the base of the OkuStim. The current of TES delivered was 150% of the IPT. If phosphenes could not be elicited during threshold determination or the participant’s IPT exceeded 0.66 mA, a treatment current of 1.0 mA was used. Generally, the subjectively worse eye, as perceived by the participant, was chosen for TES with an exception in participant 12 001 as MP could not be conducted consistently in the worse seeing eye.
Supplementary file 1
Statistical analysis
Goldmann visual fields were digitised using the Plot Digitizer V. 2.6.4 (Joseph Huwaldt and Scott Steinhorst, Massachusetts, USA). These allowed volumetric analysis of the fields using the V4e, III4e and I4e isopters to be conducted in an excel spreadsheet.18 FME (Safe Software, Ontario, Canada) converted the plots into a three-dimensional visual representation of the fields (see online supplementary figure 2). Regarding AF, the hyperfluorescent ring, characteristic of RP, was analysed according to a method previously described.19 Briefly, the hyperfluorescent ring was captured using a percentile grading system and the radius relative to the fovea was recorded over all time points. Assessments were split between the treatment period (baseline and 6 months) and observation period (between 6 and 12 months). The two periods were then compared using Wilcoxon signed rank tests due to non-normality of the data. All statistical analysis was conducted with SPSS V.22.0 (IBM, Chicago, Illinois, USA). Results are presented as mean±SEM.
Supplementary file 2
Results
Fourteen participants, aged 18–79 years, with clinically confirmed RP were recruited into an open-label observational trial from both Oxford Eye Hospital and Moorfields Eye Hospital. Seven participants had recognised mutational variants associated with RP. Participant demographics are illustrated in table 1. All 14 participants completed the initial 6-month treatment period; however, two participants withdrew during the observational period due to personal reasons (figure 1). TES was generally well tolerated with no serious adverse events (AEs) experienced during the trial supporting the primary end point of safety. A catalogue of all AEs is listed in table 2. The most frequent AEs were transient (<24 hours) self-resolving foreign body (FB) sensation experienced in two participants and discomfort underneath the skin electrode by one participant on two occasions. The latter was rectified by replacement of the electrode. The remaining AEs were not deemed to have a relationship to TES. One case of herpes zoster ophthalmicus, which affected the contralateral side, resolved with management by the participant’s primary care physician. There was also one case of supraorbital ache, although this occurred halfway through the study, was not in the location of the skin electrode and was associated with concurrent sinusitis. This resolved after 1 month with no additional treatment. Mean data and raw data values of the secondary outcome measures are shown in tables 3 and 4 respectively.
A list of all AEs encountered during the study
Mean data for all participants at baseline, after 6 months of TES and and at 12 months
Raw data for all participants at baseline, after 6 months of TES and at 12 months
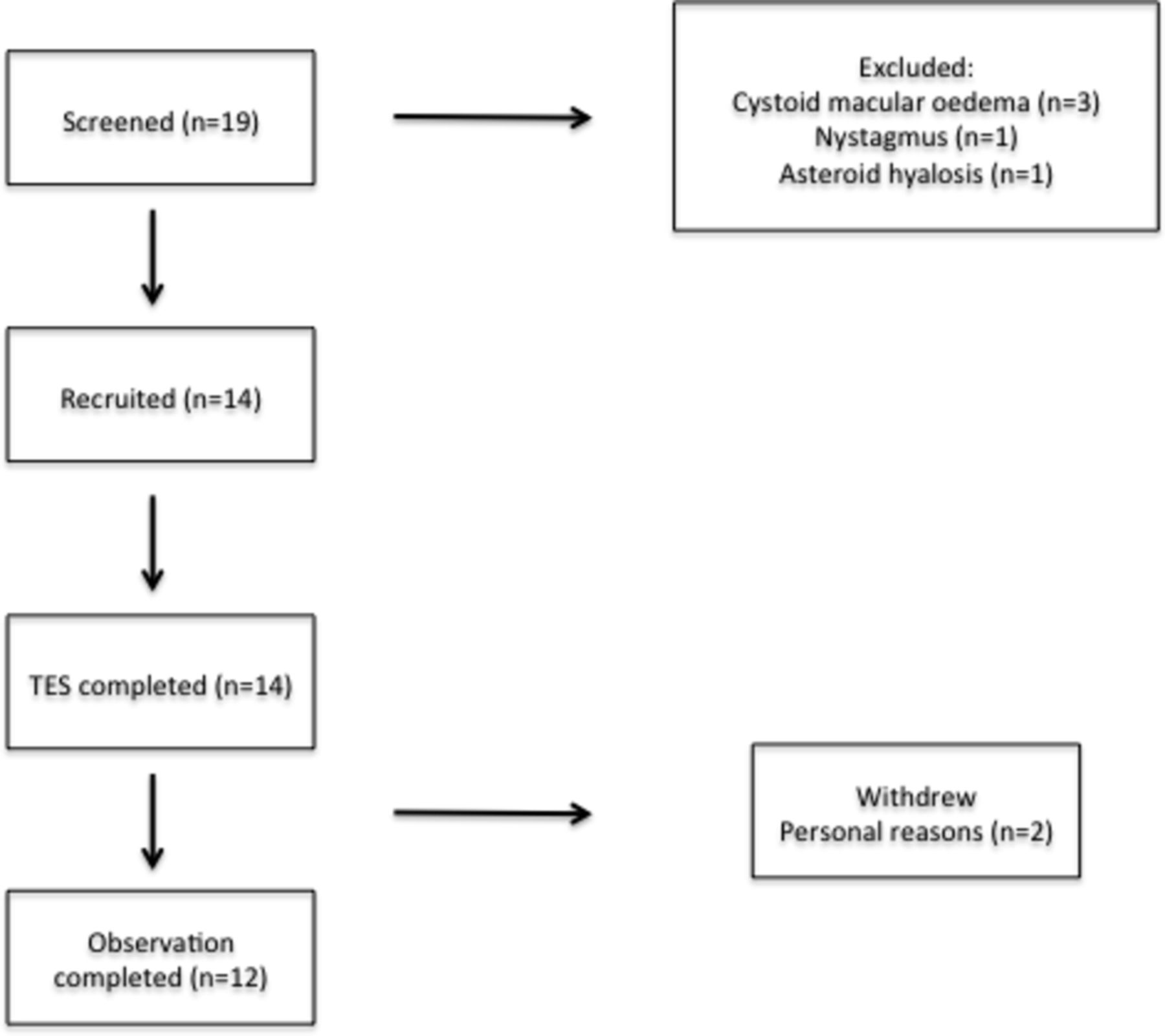
Flow chart illustrating the recruitment and progress of participants. The most frequent reason for exclusion was the presence of cystoid macular oedema (due to the independent variability in this clinical feature of RP, which would influence visual acuity readings). All participants completed the 6-month treatment period, although two withdrew during the observation period for personal reasons. TES, transcorneal electrical stimulation.
Visual acuity
Baseline BCVA was 72.3±5.9 and 71.2±4.8 EDTRS letters in the control and treated eye, respectively (n=14). Following the treatment period, BCVA increased by 1.1±1.4 in the control arm and 0.93±1.4 letters in the treated arm (P=0.58, n=14). Final BCVA (12 months following initiation of TES) relative to baseline decreased by 0.5±1.1 and 0.3±1.4 letters in the control and treated eyes, respectively (P=0.77, n=12).
Microperimetry
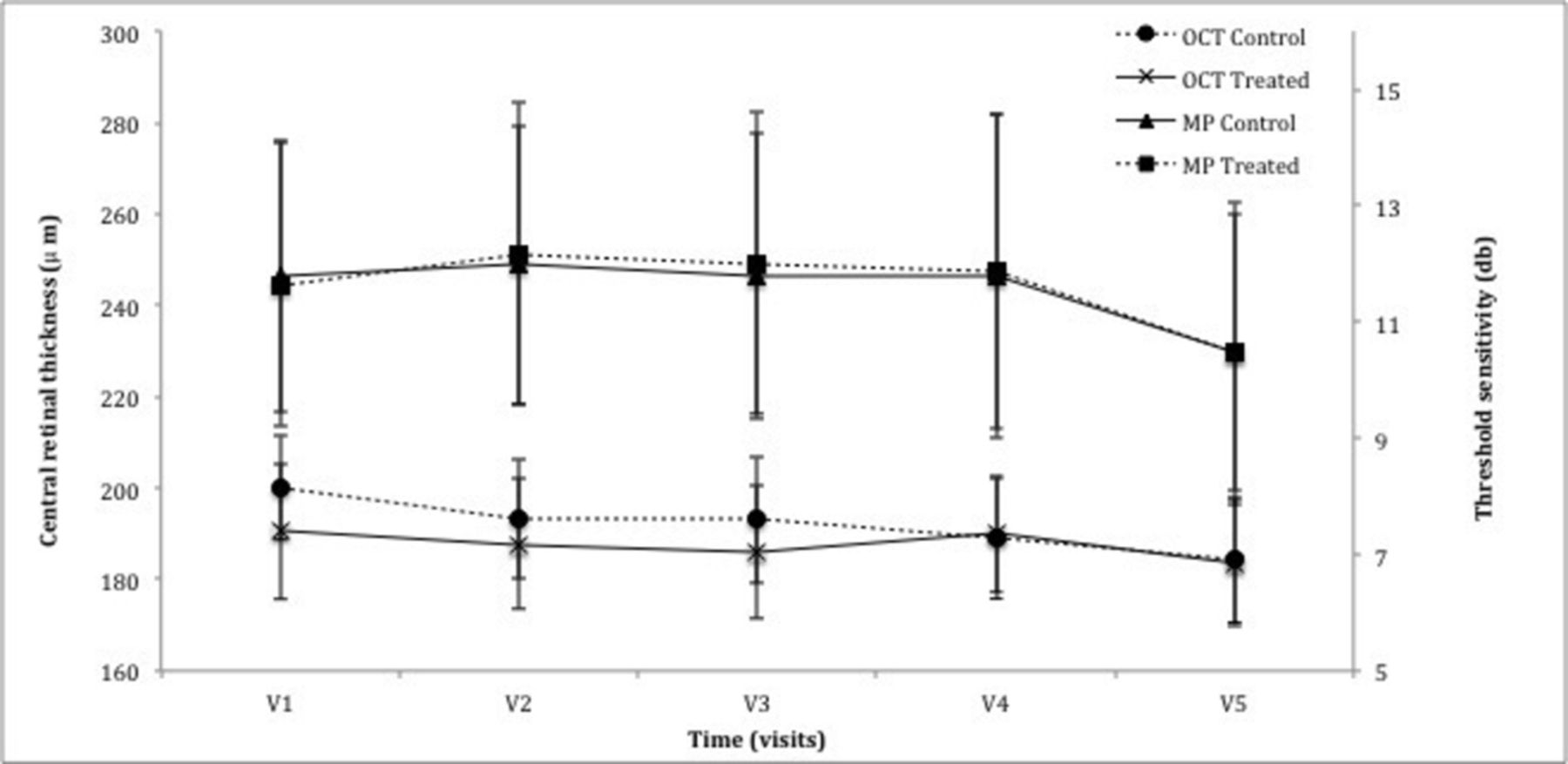
The 10–2 degree MP was carried out in all subjects, although participant P1 had poor fixation rendering the results unreliable and was therefore excluded from the analysis (figure 2). Baseline MP 10–2 threshold sensitivity was 11.8±2.3 dB for the control eye and 11.6±2.4 dB for the treated eye (n=13). At the 6-month visit, sensitivity increased by 0.02±0.51 and 0.37±0.41 dB in the control and treated eye, respectively (P=0.68, n=13). Final visit MP, relative to baseline, revealed a decrease of 0.46±0.79 and 0.56±0.53 dB in the control and treated eyes, respectively (P=0.43, n=11).

Graph demonstrating the change in central parameters of central retinal thickness (CRT) as measured by optical coherence tomography (OCT) and threshold sensitivity of the central 10 degrees using the MAIA microperimeter (MP). There was no statistical significance noted between the control eye and treated eye following the treatment period (CRT: P=0.91, MP: P=0.68) or the observation period (CRT: P=0.52, MP: P=0.43).
Goldmann visual field
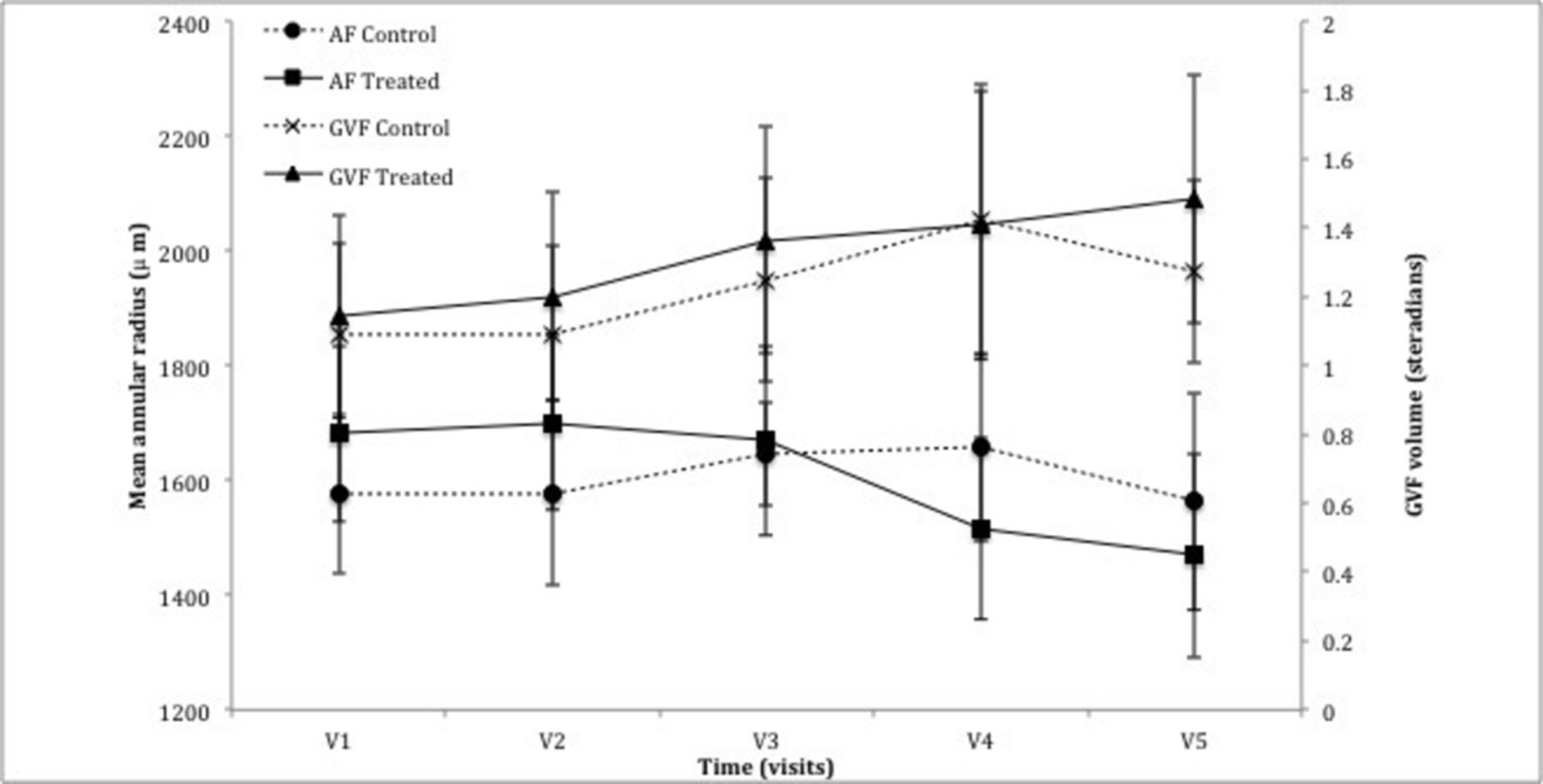
GVF analysis, as described earlier, provided a value, measured in sr. As the trial cohort consisted of participants with a wide spectrum of disease severity, GVF showed large variation with a range of 0.09–2.87 sr in the control eye and 0.10–3.43 sr in the treated eye (figure 3). Baseline GVF was 1.09±0.27 sr in the control arm and 1.14±0.29 sr in the treated arm. There was a slight increase in GVF after 6 months of TES, 0.16±0.09 sr vs 0.22±0.12 sr for the control and treated eyes, respectively (P=0.58, n=14). At last follow-up, there was a non-significant overall increase of 0.07±0.11 sr for the control arm and 0.22±0.15 sr for the treated arm compared with baseline (P=0.91, n=12).
![[SP1.jpg]](https://bmjophth.bmj.com/content/bmjophth/2/1/e000096/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
![[SP2.jpg]](https://bmjophth.bmj.com/content/bmjophth/2/1/e000096/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
{kind=link}
{kind=link}
Graph demonstrating the change in peripheral parameters of mean annular radius using autofluorescence (AF) and volume of Goldmann visual field (GVF). There was no statistical significance noted between the control eye and treated eye following the treatment period (AF: P=0.16, GVF: P=0.58) or the observation period (AF: P=0.64, MP: P=0.91).
Optical coherence tomography
Central retinal thickness (CRT) was measured using the calliper function in Heidelberg Eye Explorer software associated with the camera. On trial recruitment, four participants had mild epiretinal membranes without any traction or intraretinal fluid. One participant exhibited a staphyloma. A thickened posterior hyaloid face was noted in two participants. Cystoid macular oedema was an excluding factor and was not identified in any participant throughout the study duration.
Baseline CRT in the control and treated eye was 200±11 and 191±13 µm, respectively (n=14). At 6 months, CRT reduced marginally to 193±14 and 186±14 µm for the control and treated eye, respectively (P=0.91, n=14) with further decrease at 12 months to 184±14 in both eyes (P=0.52, n=12) (figure 2). Exacerbation of any retinal pathology, such as epiretinal membrane, was not noted through the study.
Fundus autofluorescence
As with GVF, there was a large spectrum of baseline values illustrating the spectrum of disease severity. Mean radius of the hyperfluorescent annulus at the initial visit was 1577±141 µm for the control arm vs 1681±153 µm for the treatment arm. At 6 months, the annulus radius increased in the control eye by 70±115 µm while in the treated eye, it decreased by 12±105 µm (P=0.16, n=14). At 12 months, the diameter decreased relative to baseline in both the control and treated eye by 34±150 and 268±100 µm, respectively (P=0.64, n=12) (figure 3), consistent with anatomically progressive disease.
Discussion
The results of this prospective open-label trial achieved its primary end point providing further evidence of the favourable safety profile of TES. Participants underwent 6 months of weekly TES and AEs related to the intervention were mostly isolated to mild self-limiting ocular surface disease manifesting as foreign body sensation. One patient experienced discomfort under the counter skin electrode on two separate occasions; however, this was quickly rectified by the application of a new electrode. There were no serious AEs recorded and no deleterious impact on visual function or structural measures was seen. All participants completed the initial treatment period. Akin to retinal vein occlusion, neurotrophic upregulation has been implicated in RP-associated cystoid macular oedema20 21 suggesting a possible risk with TES; however, this was not noted through the study.
Electrical stimulation therapy has been reported as early as 1873 when Dor described the beneficial impact in a case series of patients with conditions ranging from RP to amblyopia.22 More recently, renewed interest has come from the observation that those undergoing implantation of retinal prostheses showed persistent visual improvements in areas distant from the implant.8 9 Animal models of retinal degeneration have demonstrated preservation of photoreceptor and resulting electroretinography after application of TES. The completion of a sham-controlled randomised clinical trial in patients with RP undergoing weekly TES for period of 6 weeks showed that it was safe and also revealed improvements in the scotopic b-wave amplitude and kinetic perimetry (a 17% increase in the visual field area vs a 6% decrease in the sham-stimulated group) of those participants receiving a TES current of 150% of the IPT.15 A subsequent follow-up study did not reproduce the perimetric improvements seen in the pilot study, but did demonstrate gains in the photopic ERG function suggesting an impact on the cone photoreceptor system.16 Our study differed in incorporating 10-degree MP to evaluate cone function more robustly and fundus AF to look at peripheral structure. Stimulation of one eye while the other acted as a control aimed to obviate concerns over variable rates of progression between different individuals with RP.
On the basis of previous reports, we hypothesised that participants with RP might demonstrate an improvement in their kinetic perimetry and possibly 10-degree MP. However, in contrast to the pilot study, our results showed no detectable difference in the measured efficacy outcomes within the protocol of this study. A number of reasons can be proposed for this relating to both the target disease and the methodology—first RP is slowly progressive and the 6-month duration of TES may not be a sufficient period to perceive intereye differences. This becomes particularly relevant in a novel therapy, which is more likely to slow disease progression rather than reverse retinal degeneration. Second, although inherited retinal dystrophies generally affect both eyes equally, rates of progression may be slightly asymmetric confounding subsequent analysis. This pertains especially to our study where the participant’s subjectively worse eye, and therefore possibly the more rapidly progressing eye, was chosen due to the primary outcome measure of safety. A further caveat emerges when considering the phenotypic and genotypic heterogeneity of RP—regarding the former, patients with the same mutation may progress at substantially different rates rendering comparison difficult. From a genetic perspective, the varied pathophysiological mechanisms through which different mutations may ultimately induce rod-cone dystrophy might conceivably have different degrees of response to TES or indeed neurotrophic upregulation. Finally, the possibility of some unwanted signal of TES on the control eye, a stimulation crosstalk, can also not be excluded.
The stimulation parameters used in the current study reflect those in the pilot study, which were found to be safe and demonstrated significant improvements in kinetic perimetry and scotopic electroretinography. In that study, only 150% of the IPT was found to be associated with improvement but not an increased frequency of AEs. IPTs of patients with retinal dystrophies are known to exceed those of healthy volunteers with reports that the stimulation current required might correlate with the visual acuity.23 In our study, those participants in whom the IPT exceeded the protocol range generally exhibited the greatest visual field constriction. A frequency of 20 Hz stems from preclinical work demonstrating that this level confers the greatest rescue of retinal ganglion cells following optic nerve transection in adult Wistar rats.24 25 In regard to electrode choice, several groups have used bipolar contact lens electrodes to deliver TES; however, the alternative DTL electrode has been shown to be non-inferior in eliciting phosphenes and may be more comfortable and easier to apply for patients.26 Both approaches may be associated with a superficial punctate keratitis and resulting foreign-body sensation.
Recently, the advent of molecular therapies including gene therapy and stem cell transplantation hold great promise for inherited retinal diseases; however, there are potential advantages of TES. It is a non-invasive modality mitigating concerns associated with surgery such as endophthalmitis and retinal detachment as well as gene therapy implications of systemic toxicity and oncogenesis. It is well tolerated, as seen in our study, and the electrodes have nominal cost implications. Finally, the stimulation parameter adjustment is relatively simple and participants can be trained to deliver TES independently. Indeed, after the first few sessions of TES, many participants in our cohort often opted to prepare and manage the device themselves under the guidance of a trained researcher.
To more rigorously assess the efficacy of TES, a number of amendments could improve subsequent studies. As mentioned previously, the insidious progression of RP warrants a treatment period of longer duration to conclusively determine whether TES delays progression; ideally control participants would demonstrate statistically significant progression during the study for comparison with those undergoing TES. Rather than a single eye undergoing TES, it would be prudent to include randomisation and compare sham-stimulated versus TES participants to avoid possible stimulation crosstalk. It is also plausible that certain mutations may respond more favourably than others to TES; genetic sequencing may therefore be helpful to identify possible ‘responder’ groups. Notwithstanding these limitations of our study, we find the robust safety profile reported here to justify further evaluations of TES.
In conclusion, our findings confirm the long-term safety of TES justifying its further assessment. A subsequent study evaluating its efficacy in RP would ideally have a longer TES treatment period and incorporate sham-stimulated participants as a control measure.
Acknowledgments
The authors would like to thank Alexina Fantato, Anna Rudenko, Colm Andrews and Saddaf Shaheen for assistance in trial management, Barbara Wilhelm, Reinahrd Rubow and Ida Zündorf for trial design and planning and Jonathan Moules for image analysis expertise.
References
Footnotes
Contributors The project was conceived by FG with design amendments by ARW, SKD and REM. SKW, JKJ, MP, ARW, SMD and REM contributed to acquisition, analysis and interpretation of the data. SKW and JKJ composed the manuscript, which was revised with significant input by MP, FG, ARW, SKD and REM. All authors have approved the final version.
Funding The research was funded by the Oxford Biomedical Research Centre based at Oxford University Hospitals NHS Trust and the University of Oxford and Okuvision GmbH with research infrastructure support from the National Institute for Health Research (NIHR) as a Clinical Research Network Portfolio-adopted study. Okuvision GmbH participated in the design and data monitoring of the trial.
Competing interests All authors except FG were employed by the National Health Service at the time of this work taking place. SD: has previously received support to attend educational meetings from Novartis, a Novartis grant to develop the AMD service and the employer has received funds from Novartis to allow SD to speak at educational events. REM: Director of NightstaRx, a choroideremia gene therapy company established by the University of Oxford and funded by the Wellcome Trust. The company had no role in the conduct, funding or writing up of this study.
Ethics approval The study was approved by the National Health Service (NHS) Research Ethics Committee (REC) and received institutional approval from the Oxford University Hospitals NHS Trust Research and Development Department (12/SW/0293).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data relating to this clinical trial are published. Any requests for data should be addressed to the corresponding author.