Article Text

Abstract
Objective Neovascular age-related macular degeneration (nAMD) causes damage to the macula and severe vision loss. Bevacizumab is the most cost-effective nAMD treatment. The TANDEM trial was designed to determine whether, in patients with nAMD, low-dose bevacizumab is non-inferior to the standard dose in terms of visual deterioration and whether a bimonthly regimen is non-inferior to monthly, treatment as required, regimens.
Methods This was a multicentre, 2×2 factorial, double-masked, non-inferiority randomised trial with patients considered eligible if they met the National Institute for Health and Care Excellence criteria for nAMD treatment with ranibizumab. Participants were randomly assigned to standard (1.25 mg) or low (0.625 mg) dose bevacizumab and either monthly or bimonthly review regimen. The primary outcome was time to vision deterioration, defined as reduction of ≥15 letters (three lines) during the loading phase (visual acuity scores at visits B and C compared with the initial visit A), or ≥6 letters (one line) during the maintenance phase (visual acuity scores at subsequent visits compared with mean vision at visits A–C).
Results In total 812 participants (918 eyes) were randomised into the trial. The low dose showed some evidence of being non-inferior to standard dose (HR 1.07; 95% CI 0.80 to 1.42), however, there was no strong evidence of bimonthly review being non-inferior to monthly review (HR 1.45; 95% CI 1.09 to 1.94). There was no difference in visual acuity when assessed at 9 months and no major differences in the frequency of serious adverse events or reactions between the groups.
Conclusion The standard dose of bevacizumab can be halved without compromising efficacy. Bimonthly review cannot be considered to be no worse than monthly review.
- degeneration
- macula
- vision
- drugs
- treatment medical
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Research into neovascular age-related macular degeneration (nAMD) treatment had identified that bevacizumab (Avastin) and ranibizumab (Lucentis) were equally effective treatments.
This trial examined the difference in visual acuity and adverse events between standard, low and monthly and bimonthly doses.
What are the new findings?
Our trial demonstrated that the standard dose of bevacizumab can be halved without compromising efficacy but bimonthly treatment schedule did not demonstrate non-inferiority to monthly treatment.
No differences were found in the visual acuity of participants at 9 months or major differences with reported serious adverse events between the two groups.
How might these results change the focus of research or clinical practice?
These results provide useful information for clinicians patients about the dose and frequency of bevacizumab treatment for nAMD.
Introduction
Neovascular age-related macular degeneration (nAMD) is a major UK public health issue.1 Untreated, nAMD has a poor prognosis with average visual loss of 1–3 lines at 3 months from diagnosis and 3–4 lines by 1 year.2
Introduction of the angiogenesis inhibitors, bevacizumab (Avastin) and ranibizumab (Lucentis) has transformed the prognosis for nAMD.3 A major issue for treatment is cost, with bevacizumab being much cheaper than ranibizumab,3 although a barrier to use of these agents is the requirement for monthly visits; reducing this to bimonthly would greatly ease the burden. This led the East Midlands Central Commissioning Group to commission the TANDEM trial.4
At trial inception, other trials,5–8 to investigate whether bevacizumab was non-inferior to ranibizumab were being planned; and there was already strong clinical evidence to confirm that this was probably the case.4 This trial aimed to complement these studies.
Rationale for low dose treatment arms and the issue of systemic safety
The most commonly used dose of Avastin (1.25 mg) relies on the assumption that the dose should be equimolar to Lucentis, failing to take into account the fact that Avastin has two binding sites to vascular endothelial growth factor (VEGF) whereas Lucentis, being the Fab fragment, only has one. Increasing the dose of Avastin to 2.5 mg has shown no increased clinical benefit when used to treat AMD.9 10 Moreover, diabetes research has found evidence that a lower dose may be just as effective with proliferative disease responding to an injected dose as low of 0.00625 mg (6.25 µg).11
Results from previous research had identified some safety concerns with bevacizumab5 6; however, the Cochrane collaboration has concluded that the standard dose (1.25 mg) of bevacizumab and the 0.5 mg dose of ranibizumab have similar safety profiles. However, both carry the risk of systemic side effects, most importantly of stroke,12 13 so it is desirable to give the smallest effective dose. A previous study had found that increasing the dose of bevacizumab has no added benefit10 but research into reducing the dose had not been conducted.
Rationale for the bimonthly review arms and PRN regimens
Bevacizumab is an IgG monoclonal antibody directed against VEGF and ranibizumab was derived from the Fab fragment. Whole antibodies persist for months in the body whereas fragments are cleared rapidly. In humans, the reported half-life of bevacizumab is 10 days14 compared with the reported 3-day half-life for ranibizumab in primate eyes.15 Previous research16 showed that patients monitored and treated with bevacizumab on a 6-weekly basis had similar results to the ranibizumab trials.17 18 Aflibercept, which also retains the IgG Fc portion, has also been shown to be effective when given bimonthly.19
Similar results to previous trials17 18 have also been achieved without continuous dosing. The PrONTO, study showed this with a monthly pro re nata (PRN) regimen, tailoring retreatments to morphological parameters with comparable visual acuity (VA) outcomes.20 21
This trial aimed to investigate whether a low dose (0.625 mg) of bevacizumab was not inferior to standard dose (1.25 mg) in nAMD and whether a bimonthly review interval was not inferior to a monthly review interval.
Materials and methods
Study design
We undertook a prospective, multicentre, 2×2 factorial, non-inferiority randomised trial in five UK National Health Service (NHS) hospitals.
Patients and public involvement
The team consulted patients and local charities at the outset of this research and during the trial a member of the Macular Society joined the trial independent steering committee. Once the trial has been published, participants will be informed of the results via posters displayed throughout the recruiting clinics.
Participants
Participants were adults over the age of 50 years with active nAMD who met the National Institute for Health and Care Excellence (NICE) criteria for treatment with ranibizumab.22 Participants were offered fundus fluorescein angiography (FFA) to confirm the diagnosis, which was submitted for independent assessment by the UK Network of Ophthalmic Reading Centres’ Central Administrative Research Facility (CARF). Participants had refraction at baseline to obtain their best-corrected VA. Refraction is not standard practice in the NHS and those patients whose pretrial vision improved to 6/12 (logMAR 0.3) or better, remained eligible so as not to be disadvantaged. Participation in the trial was not limited to one eye, if both eyes were eligible for inclusion at baseline then both were enrolled. If during participation the fellow eye developed nAMD, it too was eligible and received treatment as part of the trial. Participants who were inactive following a period of 6 months without treatment were able to rejoin the trial. Both fellow eyes and rejoining eyes were not rerandomised and were treated according to the original treatment allocation (both dose and review schedule).
Full eligibility criteria have been reported previously.4
Between 29 November 2011 and 15 May 2012, trial coordination transferred from Bristol Clinical Trials and Evaluation Unit to the Nottingham Clinical Trials Unit; trial recruitment was suspended during this time.
Randomisation and masking
Participants were randomly assigned (1:1:1:1) to one of four groups in a factorial design: bevacizumab standard dose on monthly review, bevacizumab standard dose on bimonthly review, bevacizumab low dose on monthly review or bevacizumab low dose on bimonthly review. A secure web-based allocation schedule was used, stratified by centre and blocked using random permuted blocks of varying size. Study participants, investigators, clinical assessors and injectors were masked to drug dose allocation throughout the trial, with the visit review masked for the loading visits which took place monthly for the first 3 months. Once randomised, it was not possible to mask the participant’s visit review schedule. Masking of dose allocation was maintained for participants and clinical staff as the pharmacies dispensed the appropriate dose to the ophthalmic clinic, in a trial labelled prefilled syringe.
Outcomes
The primary outcome was time to vision deterioration, defined as reduction of ≥15 letters (three lines) during the loading phase (VA scores at visits B and C compared with the initial visit A), or ≥6 letters (one line) during the maintenance phase (VA scores at subsequent visits compared with mean vision at visits A–C).
The loading phase primary outcome definition (reduction of ≥15 letters (three lines)), before baseline vision was established, was needed to ensure patient safety. The trial began before results of the IVAN7 9 and CATT5 trials were available; therefore, it was considered sensible for patients who experienced a greater loss of vision (≥15 letters/three lines or more) to exit the trial to standard care treatment (ranibizumab or, when it became available, Eylea).
Time to visual deterioration was chosen as the primary outcome to allow participants to remain in the trial as long as they continued to respond to their treatment, reflecting standard NHS care. Equally, it meant that centres could discharge patients according to their own discharge policies (at which point the patient became censored) which was important as clinics often struggled with capacity, and the condition for adopting the trial was that there could be no additional visits over their standard care.
Secondary outcomes were VA at 9 months after randomisation and adverse events (AEs). VA at the 9-month time point was chosen to reflect the standard treatment discharge procedures for nAMD patients at the time of the trial design and meant that the trial AEs were assessed by site investigators for seriousness, relationship to bevacizumab and possible causes. All AEs were reviewed by the study medical monitor who was masked to participant treatment allocations and the independent data monitoring committee.
Procedures
Bevacizumab was provided by the compounding pharmacy at the Royal Liverpool and Broadgreen University Hospitals NHS Trust, who undertook batch testing to establish the stability, potency and sterility of the bevacizumab after aliquoting and storage.8 Participants initially received loading injections at monthly review visits for the first 3 months. Their vision was also measured and the mean of their three VA scores became their baseline VA.
They subsequently attended clinic in accordance with their allocated visit review schedule. Retreatment decisions were made on the basis of clinical and OCT appearances at the investigator’s discretion. VA was measured bimonthly and any deterioration in vision considered to meet the criterion for the primary outcome was confirmed by a refraction and repeat vision measurement. Participants confirmed to have met the primary outcome were exited and reverted to standard NHS care.
Participants remained in the study until they either had visual deterioration that met the criterion for the primary outcome, withdrew their consent, died or their disease stabilised for at least 6 months without treatment following the initial 3 month treatment loading phase at investigator discretion.
Statistical analysis
The original sample size calculation assumed one study eye per participant, 10% annual risk of visual deterioration with standard dose bevacizumab, 10% with monthly review interval, no interaction between dose and review interval, and a non-inferiority main effects HR margin of 1.4. With 90% power and 5% one-sided alpha, a total of 304 events and 2000 participants was required.
The target sample size was revised in January 2014 to 900, based on an observed annual primary outcome event rate of 20%, and annual censoring not previously included in calculations of 16%. The non-inferiority margin and total number of required events remained unchanged.
Participants were analysed according to randomised allocation, regardless of adherence, following intention-to-treat principles, with eyes as the primary unit of analysis.
Cox regression with shared frailty was used for the primary analysis of time to vision deterioration, censoring participants who had not experienced the primary outcome at study end and accounting for inter-eye correlation, with eye as the unit of analysis. There was no evidence of any interaction between Avastin dose and visit review schedule. Accordingly, we estimated main effects of dose (low-dose vs standard-dose bevacizumab) and frequency (bimonthly vs monthly) by including terms for each in the model.23 Results are presented as HRs with 95% CIs; low dose and bimonthly review visits would each be considered non-inferior if the upper 95% confidence limit for the respective adjusted HRs did not exceed 1.4.
Analysis of visual acuity at 9 months follow-up was performed using a mixed-effect model adjusted for baseline VA score and study centre as a random effect.
We investigated subgroup effects on the primary outcome according to: (1) baseline VA (≤62, >62); (2) baseline choroidal neovascularization (CNV) size (≤4, >4); (3) nAMD lesion composition (classic CNV no occult, classic CNV and occult, occult no classic CNV). These analyses were conducted by fitting appropriate interaction terms to the primary regression model.
AEs and serious AEs were summarised descriptively by treatment group.
The trial was registered: ISTRCN95654194 and EudraCT 2009-014280-38.
Role of the funding source
The funders of the study had no role in the study conduct, analysis or reporting. The corresponding author had access to all the data and has full responsibility for the decision to submit for publication.
Results
The trial is reported in accordance with the Consolidated Standards of Reporting Trials guidelines for reporting non-inferiority trials.24
Between 3 November 2010 and 31 March 2017 (recruitment suspended between 29 November 2011 and 15 May 2012), 1405 patients were approached for trial participation, of whom 812 (918 eyes) consented and were randomised into the trial (figure 1). On trial completion, the VA data for trial participants were retrospectively audited for quality, and eleven participants (14 eyes) were excluded from the primary analysis as their data could not be verified.
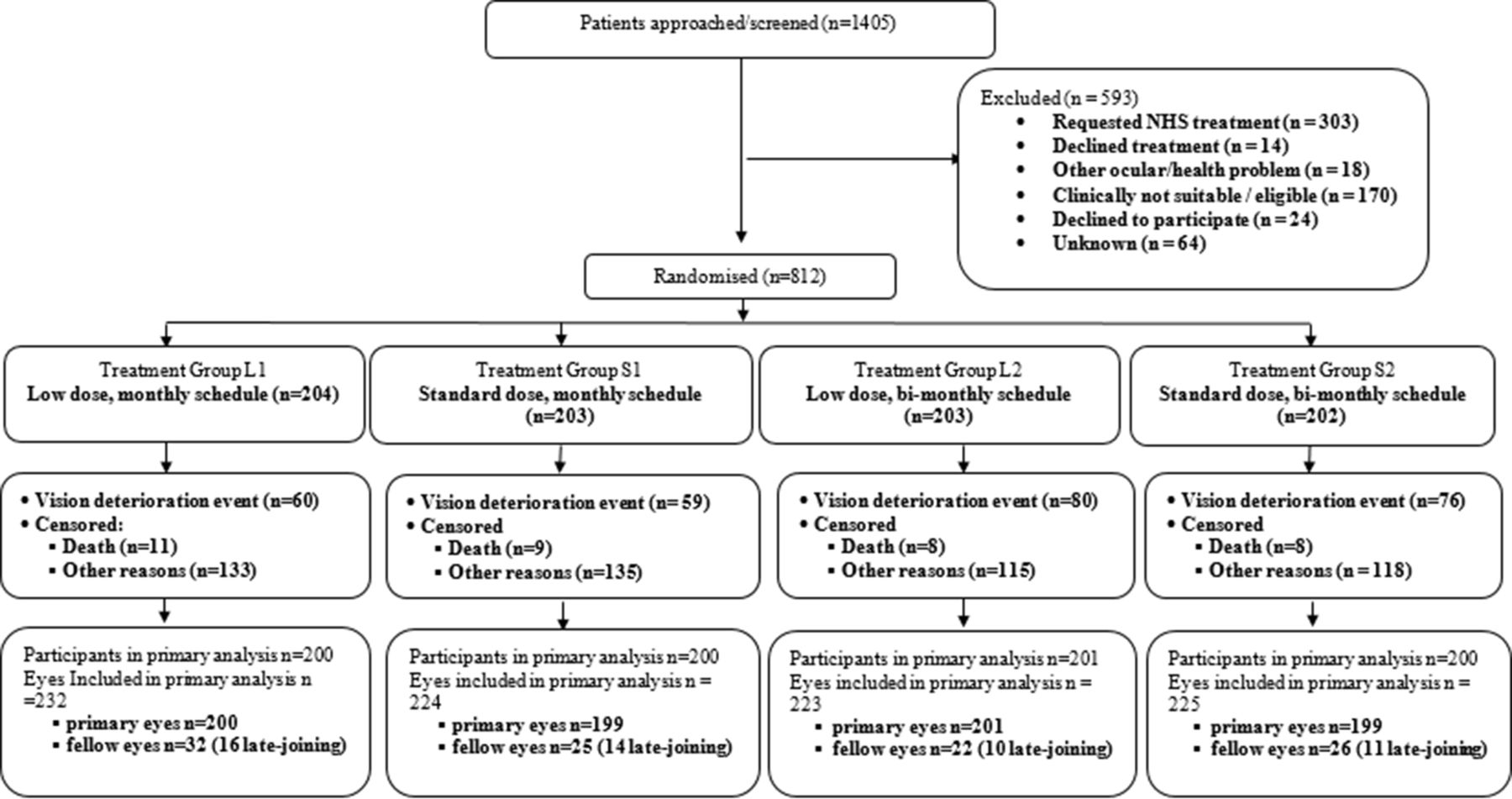
CONSORT diagram. CONSORT, Consolidated Standards of Reporting Trials. NHS, National Health Service.
Participant baseline characteristics were well balanced across the groups (table 1, online supplemental table 1). There was an equal distribution of left and right primary eyes (404 and 408, respectively) and the mean VA at entry ranged from 56.0 to 56.7 letters across the four groups. As expected, most participants were ex or current smokers (totalling 64%) with a mean participant age of 80 years.
Supplemental material
Baseline data†
Eighty-nine per cent of review visits were performed within the pre-specified time windows, (28–42 days for monthly and 56–70 days for bimonthly) with a mean between group difference of 17.35 days (35.7 days between visits for monthly and 53.05 days for bimonthly). We noted the difference of number of injections per primary eye between the two groups was approximately two (8.75 injections for monthly and 6.9 for bimonthly). Twenty-nine participants initially reported as having met the primary outcome criterion for visual deterioration were found, on review, to have their VA scores incorrectly calculated and were censored.
FFA were taken on 787 of the 918 trial eyes and these were independently assessed by CARF and agreed with diagnosis in 97%. The 22 discordant eyes remained in the trial and kept their original allocation.
Primary outcome
The number of participants with the primary outcome and reasons for vision deterioration are presented in table 2. The time to trial exit was similar for all four groups, with a median (IQR) time of 15 (9–27) months.
Primary analysis: number of vision deterioration events
The median survival time (time when half the participants are expected to experience deterioration) was 1166 days (3.2 years) for the low dose vs 1300 days (3.6 years) for standard dose and 1099 days (3.0 years) for bimonthly review vs 1321 days (3.6 years) for monthly review.
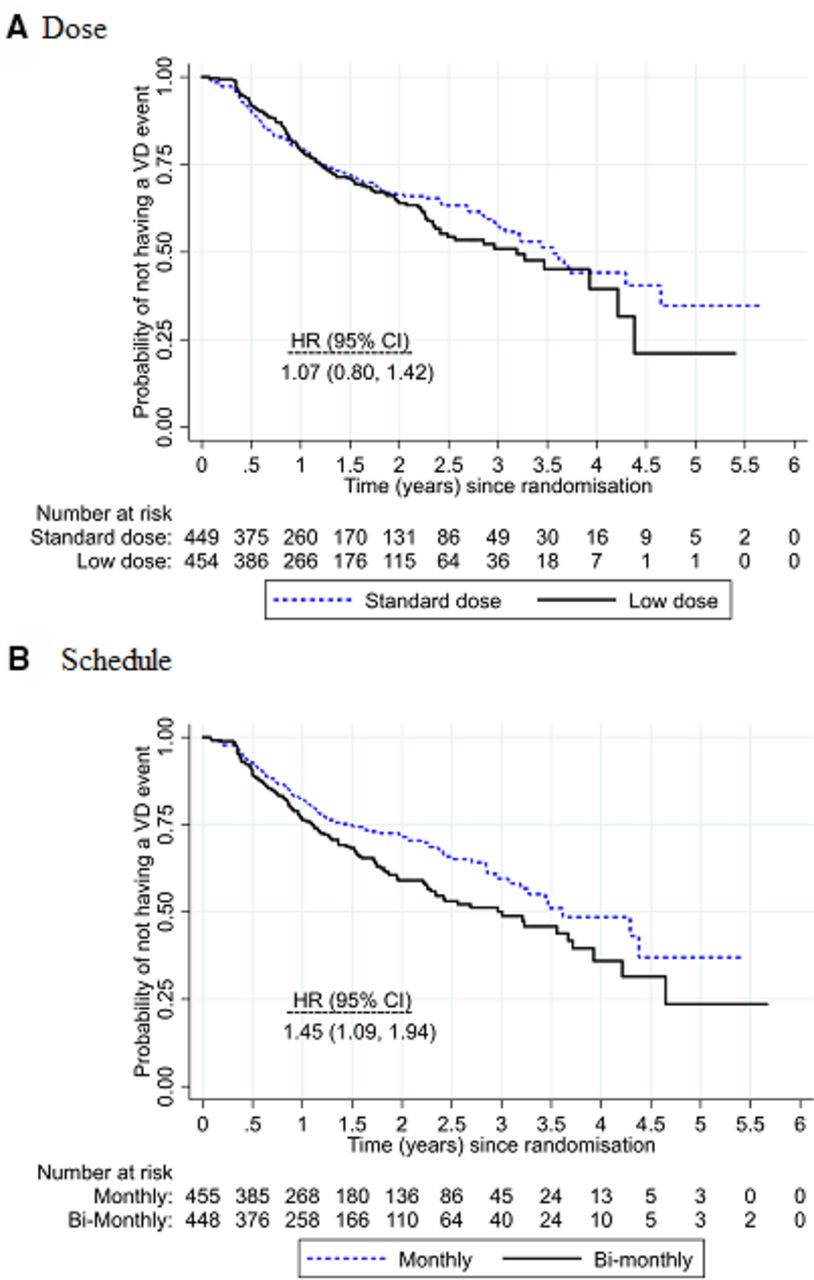
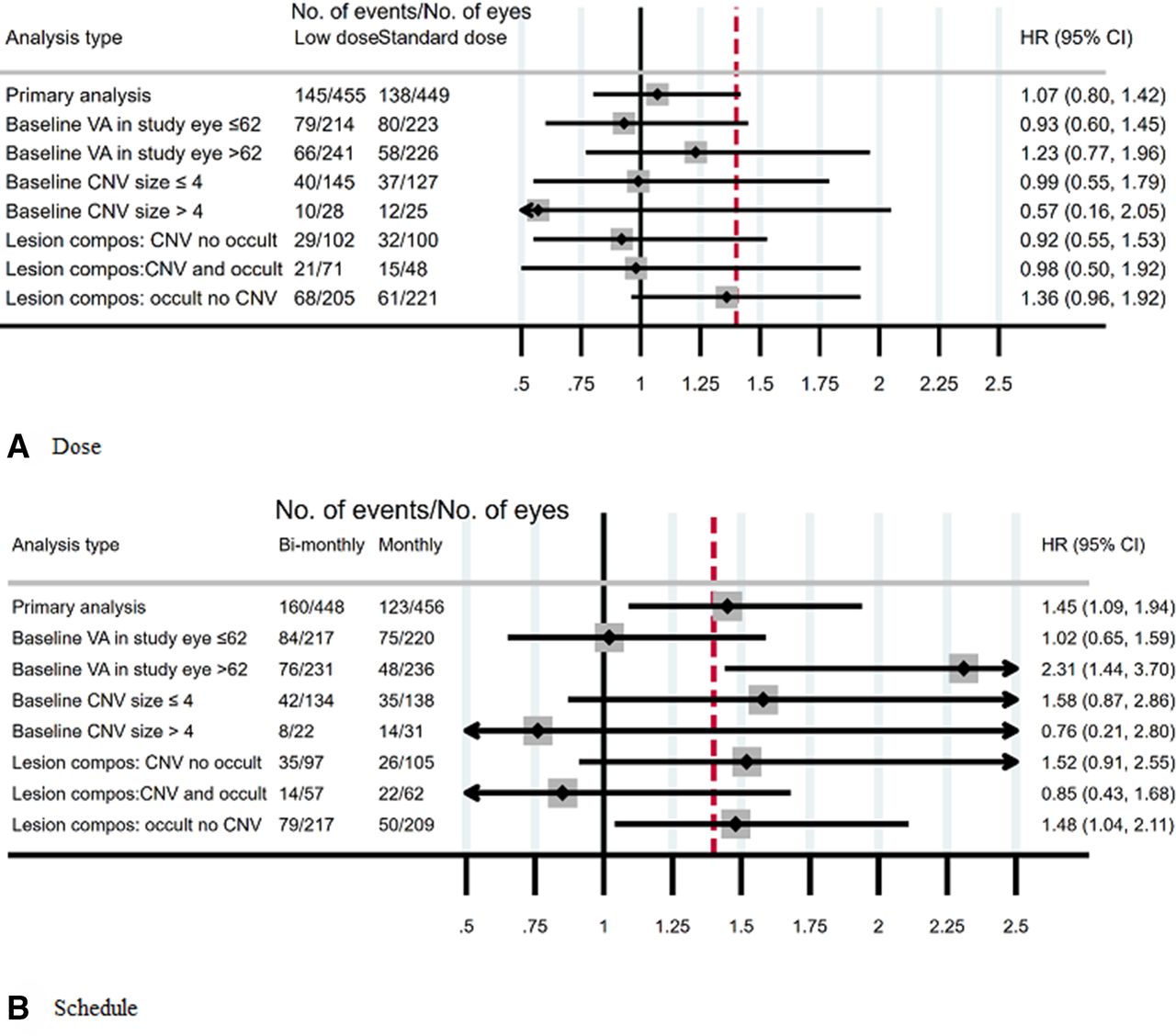
Overall, 31% of study eyes (283/904) experienced visual deterioration: 32% (145/455) for low dose compared with 31% (138/449) for standard dose and 36% (160/448) for bimonthly review compared with 27% (123/456) for monthly review. Vision deterioration was similar in the low and standard doses (adjusted HR 1.07; 95% CI 0.80 to 1.42) with upper 95% confidence limit borderline for the prespecified non-inferiority margin of 1.4 (figure 2). Vision deterioration was higher for the bimonthly review schedule compared with monthly review (adjusted HR 1.45; 95% CI 1.09 to 1.94), indicating that non-inferiority was not demonstrated. There was no evidence of any differential subgroup effects of either dose or frequency (figure 3).
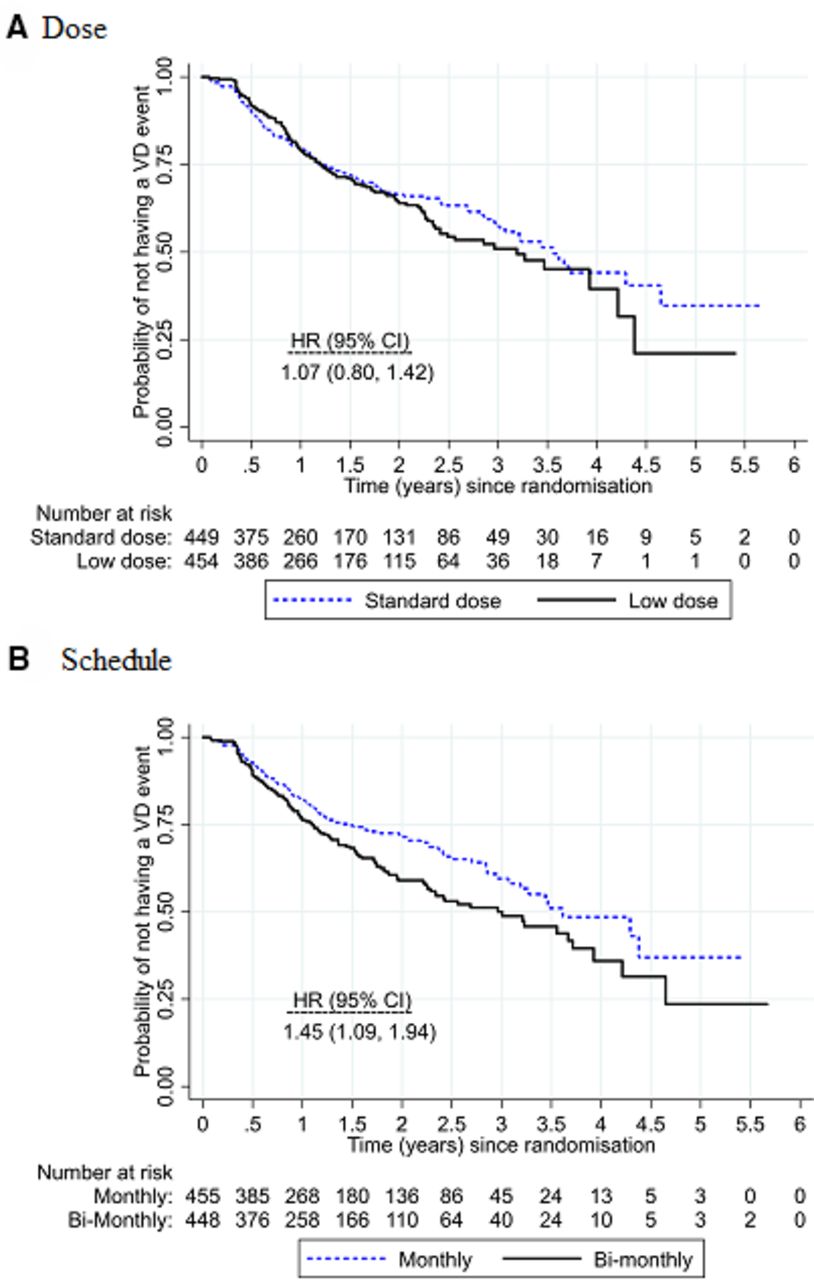
Kaplan-Meier plots of the survival curves for (A) low dose versus standard dose and (B) bimonthly versus monthly review. A table of the number at risk is shown beneath the graph. There was no evidence of interaction between dose and review schedule (interaction HR 1.31; 95% CI 0.73 to 2.36).

{kind=link}
{kind=link}
{kind=link}
Subgroup analyses for the primary outcome. Forest plots showing the adjusted HRs and the 2-sided 95% CI. All models adjusted for each intervention and study centre. The red dashed line indicates the HR=1.4 non-inferiority margin. Not all participants had the CARF data so the total number of eyes differs for each model. CARF, Central Administrative Research Facility; VA, visual acuity.
Fourteen patients lost more than 15 letters during the loading regimen; 2 in the low-dose arm and 12 in the standard dose arm.
Secondary outcomes
There was no evidence of any between-group differences in VA at 9 months, with differences in means of less than one letter (table 3). Although some participants had already exited the trial due to having met the primary outcome at 9 months follow-up and are missing from this analysis, this was similar in all groups.
Secondary outcome of visual acuity at 9 months postrandomisation
Adverse events
A large number of AEs were reported but few adverse reactions (online supplemental table 5). The only difference noted between the groups relates to more conjunctival haemorrhages and injection site reactions reported in the monthly regimen.
Discussion
The results show some evidence that the low dose of bevacizumab was not non-inferior to standard dose; the upper confidence limit only just exceeded the pre-defined non-inferiority HR of 1.4. So, in practice, the low and standard doses may be viewed as similarly effective. It was not demonstrated that the bimonthly review arms were non-inferior to the monthly review arms.
Since trial inception, a number of trials5–8 23 25–27 have demonstrated that bevacizumab is as effective as ranibizumab in the treatment of nAMD. The TANDEM trial provides evidence that low dose bevacizumab is similarly effective to standard dose. Recently, NICE concluded that there were no clinically significant differences in safety3 between the bevacizumab and other agents. We found no evidence for a difference in safety between the low dose and standard dose regimens.
We observed that the number losing ≥15 letters (three lines) of vision during the induction phase was greater in the standard dose compared with the low dose (12 cases vs 2 cases), which was unexpected. Sudden loss of three lines of vison is usually due to subretinal bleeding and it can be speculated that this is a rare consequence of blood vessel regression and so it is possible that it is causally related to use of the higher dose.
It has become clear that these treatments do not completely stabilise the disease and that vision deteriorates over time, and this was noted here. Our observed rates of visual loss were comparable to other reports.27 The SEVEN-UP study which included the original cohorts from the ANCHOR and MARINA studies showed an average of 19.8 letters lost between years 2 and 7 of follow-up which gives a rate of visual loss of about four letters per year.28 The long-term follow-up for CATT patients showed a loss of 11 letters from the end of year 2 to end of year 5 which equates to 3.6 letters per year. The rate of vision loss was significantly greater for those that had received ranibizumab compared with those who received bevacizumab.29
In this study, the median time to event (losing five letters from the average of the VA during the loading period) was 3 years for the bimonthly review arms and 3.6 years for the monthly review arms for those with active disease, equating to a difference of under half a letter per year in the rate of visual loss of between the regimens. This difference is of uncertain clinical significance and both monthly and bimonthly as required (PRN) regimens can be considered effective in the treatment of nAMD; the more intensive (monthly review regimen) may be appropriate for patients at a critical threshold where the loss of a couple of letters would be significant. However, some patients may accept a slightly worse outcome as they prefer a less intensive review regimen.30
The median number of injections per participant was 5–6 (online supplemental table 4), which was fewer than in other studies and yet the results were comparable, suggesting that there may be a tendency to overtreat. The FLUID study showed that subretinal fluid could be safely observed in selected cases, showing that it is not always an indicator of active disease.31 Here, it was observed that there were more macular haemorrhages causing severe visual loss in the standard arms during the induction phase. This was an unexpected observation but suggests that in some cases, haemorrhage may be due to blood vessel regression and not due to disease activity.
A particular advantage of the PRN regimens is that it allows early recognition of inactive disease thereby preventing over treatment. A disadvantage is that those with active disease may get under treated. A compromise position is to consider treating those with active disease with a ‘mini-course’. The PRN regimens in the CATT study5 6 did slightly less well than those given monthly injections where a single injection was given if there was disease activity. In the IVAN PRN group, retreatment when required was given by a course of 3 monthly injections, and there was no difference noted between the PRN and monthly groups.7 8 The VIEW 1 and 2 studies showed that aflibercept was not inferior to ranibizumab17 32 and can be given bimonthly on a continuous basis.
Strengths of this study were the inclusive nature of the study population and the length of follow-up. As a pragmatic trial, TANDEM included all patients who met the criteria for standard care anti-VEGF treatment. Patients who declined to have a FFA but were still thought to have nAMD were still eligible for enrolment into TANDEM; 86% did have a pretreatment FFA and in 97% of these, the diagnosis was confirmed by CARF.
All previous trials have been limited to 1–2 years which meant that long-term visual deterioration could not be observed. As a time-to-event trial, participants remained in the trial with active disease for up to 5 years, allowing assessment of the long-term outcomes for the majority of participants. In contrast, there were a subset of participants (n=176) who could not be followed to vision deterioration or stable disease as they remained disease active at the point the trial ended.
The trial had limitations. The time-to-event trial design is unusual for nAMD trials and has made it difficult to directly compare results with previous research. The design was chosen for reasons of patient safety; it enabled anyone with visual deterioration to be exited early from the trial back to routine NHS care. This was appropriate at the time of trial design since the trials demonstrating non-inferiority of bevacizumab had not been reported.
The trial was designed to run in parallel with routine NHS care, and all services were (and are) struggling with capacity. This meant that centres had to discharge patients who were thought to have inactive disease and so the long-term results are in terms of those with ‘active disease’ rather than the total population.
This study has shown that although the upper confidence limit just exceeded the predefined non-inferiority HR of 1.4, low and standard doses might in practice be viewed as similarly effective in the treatment of nAMD.
The study did not provide evidence that bimonthly PRN regimen is non-inferior to monthly. Continuous regimens run the risk of over treatment and PRN of under treatment. NICE modelled the existing regimens and concluded that there was little difference between any of the commonly used ones; but this remains an area for future research.
Research in context
TANDEM is the UK’s largest nAMD clinical trial (and one of the largest conducted worldwide) with the participants accurately reflecting the NHS patient population. TANDEM was unique in that it was embedded within ophthalmology clinics and was the vehicle for delivery of routine NHS care. This design allowed for prolonged observation of participants of up to 5 years, exceeding previous trials where follow-up typically ended at the 1 or 2 year time point. This is particularly relevant as it is after 2 years of treatment that visual outcomes have been shown to drop off.
TANDEM is the only trial to date that has directly investigated whether a bimonthly bevacizumab regimen is non-inferior compared with a monthly treatment. This has been given particular relevance recently as bimonthly bevacizumab is the only nAMD treatment regimen that met the NICE criteria for cost-effectiveness on their health economic model (though they have approved other regimens).
Acknowledgments
We thank NHS England and the following Clinical Commissioning Groups for providing funding: Nottingham City, Nottingham West, Nottingham North and East, Rushcliffe, Mansfield and Ashfield, Newark and Sherwood, Mid-Notts, Leicester City, Lincolnshire Community Health Services NHS Trust, North Derbyshire. Thank you to the independent members of the trial steering committee, who provided independent oversight and the independent data monitoring committee: Greg Fell (Chair, TSC), Cathy Yelf, Philip Lubert, Richard Wormald, Catey Bunce, Adnan Tufail (Chair, DMC), Gary Collins and Hugh McIntyre. The following members of site staff: King’s Mill Hospital (Manju Mukherji, Karen Meikle, Ruby Amankwah); Lincoln County Hospital (Bola Ajimoko, Bronwen Attrup, Ann Marsh); Leicester Royal Infirmary (Konstantinos Tsaousis and Mandy Babbington), Queen’s Medical Centre (Alison Fotheringham, Mary McKeating and Emily Hogan). Thank you to the Bristol Clinical Trials and Evaluation Unit who initially designed the trial. We want to thank you the many staff, based at the Nottingham Clinical Trials Unit, who contributed to the trial over its nine year duration and finally, and most importantly, the trial participants.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors AF was the chief investigator and conceived the idea and drafted the manuscript. The protocol was developed by AF in conjunction with the team at Bristol Clinical Trials and Evaluation Unit. RH managed the trial (January 2015 to close), with oversight from MC (February 2016 until March 2016) and EM (March 2016 to close). MO and RO undertook statistical analyses, with support from AAM. TE, SD-M and PT were principal investigators at recruiting sites and recruited patients into the trial. LMD was responsible for oversight of the NCTU trial coordination. All authors contributed to revisions of the trial manuscript and approved the final version.
Funding The study was initially commissioned by the Primary Care Trusts and then by the local Clinical Commissioning Groups when commissioning for the NHS was reorganised. At the time of the reorganisation, NHS England agreed to meet the costs associated with image grading.
Competing interests AF was on the NICE AMD guidelines and the NICE serious eye disorders quality improvement committees.
Patient consent for publication Not required.
Ethics approval The trial was approved by the Leicestershire, Northamptonshire and Rutland Research Ethics Committee 1 (ref 2009-014280-38).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon request - data will be avaliable a minimum of 12 months following publication and all data sharing requests must be reviewed by the data sharing committee.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.