Article Text

Abstract
Proboscis lateralis (PL) is a rare congenital malformation of the craniofacial structure with varied clinical associations. None of the studies documented a discrete review of ophthalmic presentations in PL. The principal aim of the present study is to explore the ophthalmic manifestations of PL. The ancillary goal is to derive a relationship between congenital deformity in PL and various ophthalmic anomalies. Databases were searched in order to obtain articles related to PL. A qualitative systematic analysis of 100 subjects was performed. In PL, eyelid coloboma (32.6%) is the most common ocular feature, followed by hypertelorism (25.3%), iris coloboma (22.4%), lacrimal system abnormality (20.7%), malpositioned eyebrow (14.4%) and retinochoroidal coloboma (12.9%). Sinonasal deformity is the most common systemic abnormality, detected in 87.9% of cases of PL, as compared with central nervous system involvement (56.2%) and other anomalies. The analysis showed a strong significant association between brain abnormalities and hypertelorism (p=0.000) and between brain abnormalities and micro-ophthalmia/anophthalmia (p=0.000). Statistically significant association was noted between cumulative ocular abnormalities and cumulative systemic abnormalities (p=0.001). The present study on PL reviewed the salient features of this rare congenital disorder. The study outcome provides a new aspect to concomitant ocular abnormalities. This study supports the view that other congenital anomalies in cases of PL had significant influence on certain ophthalmic anomalies.
- embryology and development
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Proboscis lateralis (PL) is a rare, congenital, off-centre, vertical deformity characterised by a rudimentary nose-like structure. The documented occurrence is less than 1 per 100 000 population.1 It is embryologically related to a facial fusion defect. The tubular process often has a unilateral presentation and projects from the inner corner of the orbital roof.2 It measures around 2–3 cm long and 1 cm in diameter. The structure may include all the tissue lines analogous to a normal nose. Sometimes a patent tract transverses the entire proboscis exuding discharge.3
The first case of PL was described in 1861 by Forster,2 and many reports have followed afterwards. Of interest to an ophthalmologist are the concomitant ocular anomalies reported at around 44%–70%.1 4 The spectrum of ocular abnormalities includes hypertelorism, anophthalmia, microphthalmia, abnormal lacrimal system, microcornea, lenticular opacities, coloboma eyelid, coloboma iris and retinochoroidal coloboma. Less common are cyclopean eye, hypoplastic orbit, ptosis, corneal opacity, etc.4 5 The literature on this ophthalmic condition is sparse given its rare occurrence. There are only four cases published in ophthalmic journals: two were published in the Archives of Ophthalmology2 6 in 1947 and 2001 and the other two in Ophthalmic Plastic & Reconstructive Surgery7 8 in 2008 and 2016. The primary aim of this review was to study this rare malformation from an ophthalmic perspective, and second to derive a correlation between ocular and associated systemic malformations.
Methods and materials
Patient and public involvement
Patients were not involved directly or indirectly from inception of research to the final documentation for readability or accuracy.
Study sources and selection
We identified publications pertaining to PL and reviewed cases of PL with a retrospective chart. Investigators collected cases recorded between January 1885 and November 2019. Cases were evaluated and the data were compiled.
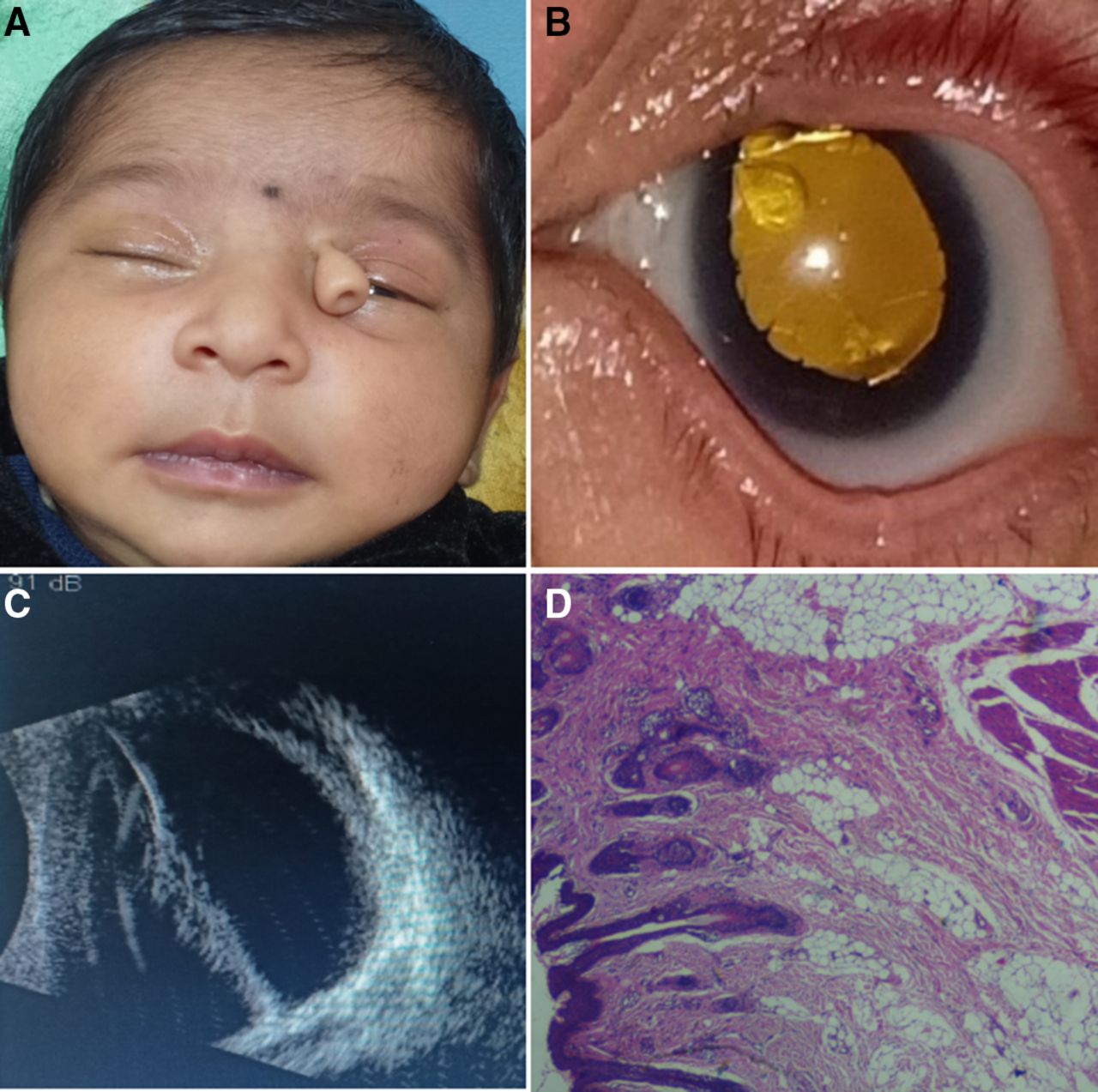
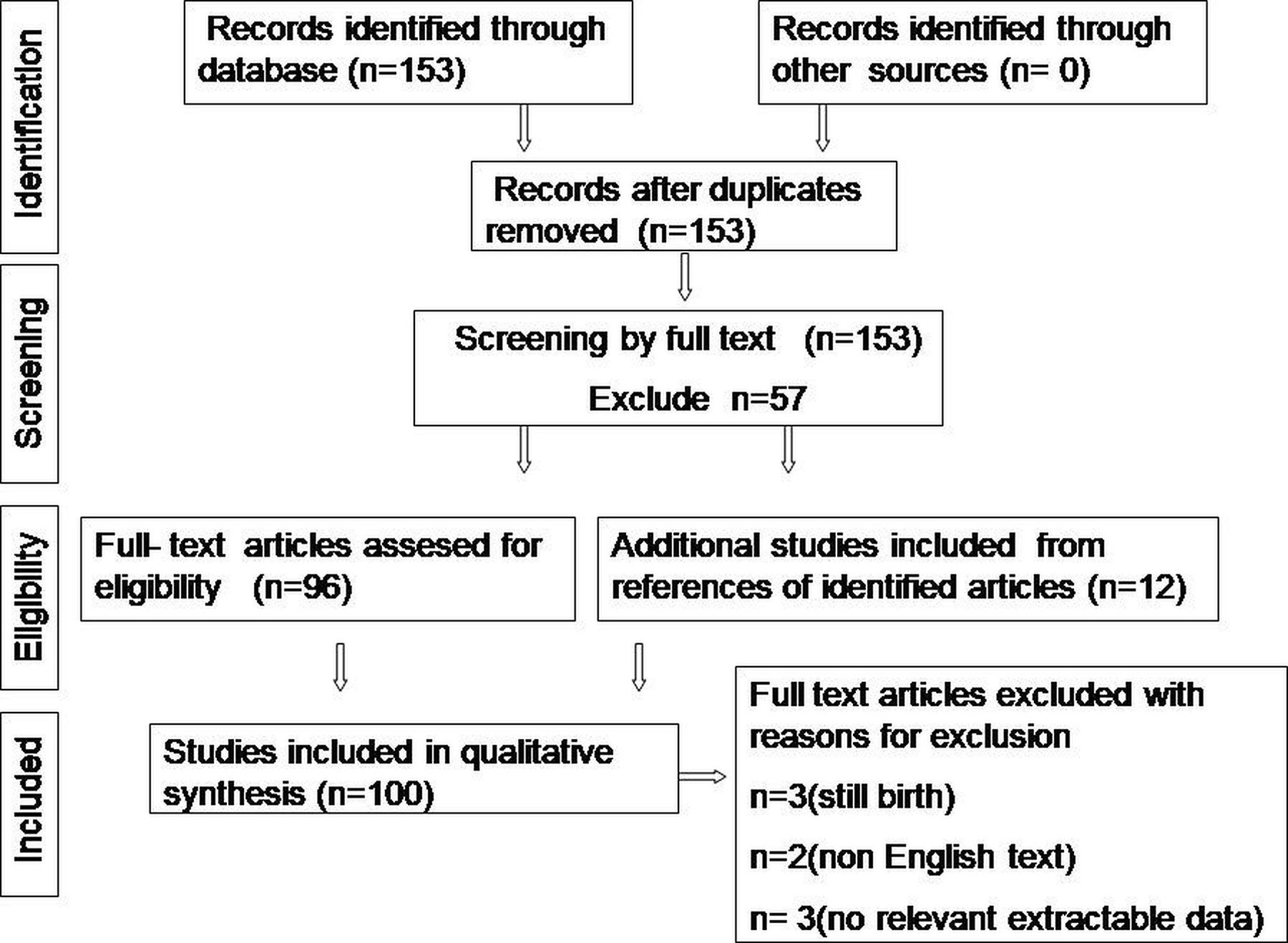
An electronic-based systematic literature search was performed in PubMed, PubMed Central, Cochrane Library, ResearchGate and Google Scholar. Search keywords included ‘proboscis lateralis’ and/or ‘supernumerary nostril’. Filtered database search revealed 153 case reports/case series according to their titles. From the 153 publications, we identified a total of 97 articles with full-text manuscripts. Full text was assessed for eligibility independently by two authors. The reference lists of published cases obtained during this phase were also screened. Twelve relevant articles were further identified. Nine cases were later excluded based on the following: (1) non-English articles, (2) reports with stillborn or those who did not survive long enough to undergo an ocular examination, and (3) reports with no mention of ocular examinations or extractable ocular data. We identified 100 cases, including one of our own (figure 1) which met our research criteria for analysis. The search strategy and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses flow chart illustrating the process are presented in figure 2.

(A) Full face of an infant showing left-sided proboscis lateralis and normal nose. (B) Anterior segment photo showing abnormally shaped cornea, corectopia, persistent pupillary strands, large Mittendorf dot, lenticular opacity and yellow fundal glow. (C) B scan ultrasound showing echogenic stalk extending from the back of the lens towards the retinal surface suggestive of persistent hyperplastic primary. (D) Histological section showing the stratified squamous epithelium with adnexal gland overlying the mixture of mature fibrolipomatous tissue, striated muscle fibres and blood vessels (H&E ×10 magnification).

{kind=link}
{kind=link}
PRISMA flow chart illustrating the process of selection of case records for systematic review. PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Data extraction
Data were extracted from each case and further processed. We collected the demographic characteristics, ocular findings and systemic manifestations of each patient. The present study focused on ocular anomalies under two subdivisions: adnexa and intraocular manifestations. The adnexa included the orbit, eyebrow, eyelid, conjunctiva and extraocular muscles. The intraocular subset included the rest of the ocular structure. Micro-ophthalmia or anophthalmia evaluation was carried out as a separate entity. Besides the documented adnexa abnormalities, facial photos were scanned to elaborate on the details of the adnexa. The intraocular abnormalities reported were the ones included and accounted. The rest of the intraocular findings were assumed normal if not specified. Information collected on the associated systemic anomalies was categorised into sinonasal abnormality, oroclefting, facial deformity and other systemic malformations.
Further, patients were classified into four groups as per the Boo-Chai’s classification4 for convenience of comparison with other series. A scoring system is formulated to measure the extent of ocular and systemic abnormalities for each subjects. A score of 1 is allocated to each abnormal ocular and systemic finding. These points are then summated to provide a total scores which draws up the distinctive cumulative ocular and cumulative systemic score reflecting extent of abnormalities for each subject. The higher the cummulative score the more abnormalities. The scores aid in deriving association between ocular and systemic abnormalies as well as comparison among various study attributes. The cumulative ocular score was further allocated into complete adnexa score and complete intraocular score for subanalysis.
Statistical analysis
Data were analysed by SPSS V.7.0 software using descriptive and inferential statistics. Results for continuous variables were presented as mean±SD. Categorical results were presented in numbers (%). We assumed that the observations recorded for a continuous variable had followed a normal distribution. Pearson’s χ2/Fisher’s exact tests were used to investigate the association between the selected variables. A probability value of p=0.05 was considered statistically significant, while values of p=0.01 and above were considered strongly significant.
Results
Given the retrospective nature of the present study, the computed results have excluded missing variables. Gender was reported for 88 patients, 53 of whom were male. The male to female ratio was 1.5, against 2:1 reported in the literature.4 The average age at clinical review was 2.45±4.85 years, ranging between 0.0 (at birth) and 34.0 years. In 81 cases PL was positioned unilaterally, in 3 cases midline, and 6 reported cases of proboscis-like nares that did not follow any laterality. Literature reports bilateral PL9; however, these cases did not meet the inclusion criteria for the present study and hence excluded. Out of 100 cases, 9 had an atypical position on the cheek,10 chin,11 ala of the nose,12 upper lid12 and lateral canthus.13 Of 41 cases, discharge from PL was present in 26 cases (63%).
Ophthalmic structural abnormalities
The three most common ocular abnormalities in PL were eyelid coloboma (32.6%), hypertelorism (25.3%) and iris coloboma (22.4%).
Among all eyelid colobomas, lower eyelid involvement was recorded in 21 (67.7%) cases, upper eyelid in 8 (25.8%) cases, and both the upper and lower eyelids were involved in 2 (6.45%) cases. Other observed eyelid abnormalities were fissure asymmetry in five eyes, ptosis in three eyes and epicanthus in two eyes.
Other ophthalmic anomalies included were lacrimal system abnormality (20.7%), malpositioned/abnormal eyebrow (14.4%) and retinochoroidal coloboma (12.9%). Micro-ophthalmia/anophthalmia was observed in 20 cases (20%), of which 13 (65%) were unilateral and 7 (35%) were bilateral. Table 1 presents a summary of the remaining less prevalent ocular abnormalities.
Observed complete ocular features in cases of proboscis lateralis
Recorded rare presentations with each case were dystopia, lagophthalmia, malposition of the orbit, nystagmus, conjunctival cyst, abnormally shaped microcornea, corectopia, persistent iris strand, persistent hyperplastic primary vitreous, peripapillary staphyloma, optic disc pit, telecanthus, choroidal cleft and retinal detachment.
Systemic associated abnormalities
The most common systemic abnormality associated with PL was sinonasal deformity with 87.9% (87 of 99), followed by central nervous system involvement with 56.2% (18 of 32), facial deformity with 54.7% (29 of 53) and cleft lip/palate with 24.0% (24 of 100). Table 2 shows the systemic abnormalities with predominance of their subtypes. The frequently observed subtypes comprised heminasal aplasia (53.5%) and ethmoidal hypoplasia (22.2%) among cases of sinonasal deformity, high-arched palate (15.1%) among facial deformities, and ventricular dilatation (34.4%) among brain abnormalities. Out of 32 cases, the less common systemic association included genital hypoplasia, which was found in two cases, and one case each of kidney agenesis, single ureter, ventricular septal defect and patent ductus arteriosus. Infrequently detected anomalies included inguinal hernia, single umbilical cord, absent premaxilla and abnormal pyriform.
Assessment of systemic abnormalities of proboscis lateralis
The results of the analysis of the classified groups are as follows: group 1 of PL with a normal nose was the least prevalent (11.2%); group 2 was associated with only nose deformity, with a prevalence of 18.4%; group 3 involving the nose and the eyes and/or adnexa abnormalities was the most common (46.9%); and nasal and ocular defects along with cleft lip and/or palate comprise group 4 (23.5%). These results support the observation made in Khoo and Boo-Chai’s study,4 where group 3 was the most common and group 1 was the rarest.
Associations between ophthalmic and systemic abnormalities
The statistical agreement showed the significant association between cumulative ocular abnormalities and cumulative systemic abnormalities (p=0.001). Yet an association between cumulative ocular abnormalities and brain abnormalities was insignificant (p=0.249). Further analysis establishes a significant relationship between brain abnormalities and complete adnexa findings (p=0.018). No statistical association was found between complete intraocular findings and brain abnormalities (p=0.991). Hypertelorism and micro-ophthalmia/anophthalmia showed a significant association with brain abnormalities (both p=0.000) (table 3).
Associations between brain abnormality with adnexa hypertelorism and micro-ophthalmia/anophthalmia
Online supplemental table 1 provides a comprehensive chart of ocular and systemic findings in 100 patients.
Supplemental material
Discussion
Ophthalmic manifestations in PL have unsatisfactory documentation in the past, either in small series or case reports. However, combined data offer an approximate prevalence of ocular abnormalities.
A summary of embryonic development is necessary to understand the genesis and pattern of systemic malformations and its relationship with ocular abnormalities. Head and face development occurs between 4 and 8 weeks of embryogenesis. The insult during this period results in a constellation of malformations. However, the causative factor is uncertain.14
The development in early embryo occurs via series of orderly induction. Embryonic induction is a process by which a group of cells called ‘organiser’ influence and differentiate the adjacent embryonic cells. This is a recurring phenomenon and an array of organisers are produced in an orderly progression, from a ‘primary organiser’ to secondary and tertiary organisers, and so on. 'Each order of organisation results in a particular development abnormality'. The interaction between the inducer and the tissue being induced is probably chemically mediated. The extent of interaction is limited by finite distance and critical exposure time within a field volume. The influence of organiser is mostly found at the centre of the field volume and least at its periphery. An overlap from the surrounding organisers at the field perimeters may result in small or otherwise malformed structure.
The notochordal process is the first and the most important organiser in the head of the embryo. It induces formation of neural tube and foregut, which act as secondary organisers. The neural tube is closed by 4 weeks of gestation and a primitive forebrain (prosencephalon) emerges at its rostral end. Neural crest-derived mesenchyme from the prosencephalic region forms the unpaired central frontonasal process. During weeks 5 and 6 of gestation, a series of cleavages in the prosencephalon induce the neuroepithelium placodes in pairs (optic, otic, olfactory in the same order). Similarly, the foregut organises the formation of the maxillary process from the first branchial arch. The primary defect of ventral induction during prosencephalon development results in the most profound anomalies, holoprosencephaly. In holoprosencephaly, various states of failure of differentiation occur, including improper placement of the interplacode area. As a result, the central proboscis and other severe anomalies such as cyclopia, midfacial clefts and severe hypotelorism develop.15
The olfactory placode is the primary organiser of a developing nose. At week 5 of gestation, the nasal groove is formed from the olfactory placode. It interacts with a frontonasal process to define the maxillary process, the medial nasal process and the lateral nasal process. During weeks 6–7, the maxillary process induces transformation of the nasal and oral cavity. The medial nasal prominences merge with each other across the midline and interact with growing maxillary processes to form the intermaxillary segment. The ventral end of the median nasal process extends into the mesenchyme of the roof of the mouth. This enlarged area is identified as globular process. The interaction between maxillary process, lateral nasal process and the intermaxillary segment ultimately forms the upper lip, the zygomas, the maxilla bilaterally, the philtrum and the nasal bridge by week 10. The primitive anterior nares, mouth and alveolus form when the maxillary process meets the globular process. For all obvious reasons, the maxillary process is key to the development of facial structures. The maxillary process regulates and gets regulated by the optic, otic and olfactory centres.15 Hence, any localised facial defect may harbour maxillary maldevelopment and vice versa.
During expansion of the medial nasal prominence, a fissure may develop, leading to two abnormal fragments of the prominence. The abnormal lateral fragment merges with the lateral nasal prominence and forms PL. The medial fragment, which merges with the unaffected side, is destined to another fate. This deviation results in a PL with abnormal nose and an additional hypoplastic maxillary prominence can have various associated abnormalities. Alternatively, an extra nasal placode when arranged in a vertically stacked manner on the affected side forms a PL with a normal nose. The lower placode contributes to formation of a normal nose, while the upper placode remains isolated and develops into a PL.14 16
Development of eye and palate gets affected when the inductive influence on their adjacent fields deviates. Eye development begins with optic placode formation from the neuroectoderm at week 3 of embryogenesis. Around week 5, auto-invagination in the optic vesicles creates a double-walled neuroectodermal optic cup. At the same time the optic vesicles interact with the surface ectoderm to form lens placode, which transforms into future lens. The connections of optic vesicle to the forebrain attenuate to form optic stalks with a groove on their inferior surfaces. This groove is referred to as optic or choroidal fissure and closes by week 8. Inadequate closure of the optic furrow results in coloboma of the iris, retina and disc.14 The interplay between the maxillary process and other developing mass shapes the extraocular structure. At week 7, the maxillary process and the lateral nasal process interact to form the lacrimal apparatus. The nasolacrimal duct forms when plugged epithelium recanalise at week 24. The mesenchyme along the optic vesicle and maxillary process contributes to the formation of medial aspect of the lower eyelid.
The line of interaction between various processes creates grooves which eventually obliterate between weeks 7 and 20.15 Failed fusion between the maxillary process and the undifferentiated facial prominence leads to various degrees of embryonic fissure defect, facial dysmorphogenesis, midfacial hypoplasia, orofacial clefts, intercanthal defect, sinuses hypoplasia and many more. The underdevelopment of the maxillary process also affects the medial migration of the eyes from 160° to a final position of 72° between the optic axes.14 The retarded maxillary growth produces some degree of hypertelorism.
Our finding of 73% prevalence of ocular abnormalities in PL is consistent with that reported by Khoo and Boo-Chai (70.5%).4 On the contrary, English17 noted a lower prevalence (44.0%) of ocular abnormalities.
Eyelid coloboma represents the most common ocular abnormality in this study. The present study showed higher involvement of the lower eyelid (67.7%). Our result is at odds with studies18 concluding more of upper eyelid involvement (93%) in congenital eyelid colobomas. These studies included both isolated colobomas and colobomas coexistent with other craniofacial anomalies. The latter has its origin earlier in the embryogenesis. Craniosynostosis syndrome and cleft disorders contributing to lower eyelid colobomas were under-represented. This introduces a probable source of discrepancy with the present results.
The incidence of ocular coloboma reported in ophthalmic literature is 36% for the anterior segment, 39% for the posterior segment and 24% for both the anterior and posterior segments.19 The frequency of iris coloboma (22.4%) and retinochoroidal coloboma (12.9%) in our study is much lower. Expertise bias while assessing for ocular anomalies at a younger age in the present cohort may explain the difference.
Hypertelorism documented in the current study is lower than that reported by Sakamoto et al20 (25.3% vs 54%). Illusory hypertelorism without radiographic or CT confirmation is difficult to rule out. Thus, hypertelorism adopted in present study was the one reported by respective authors of case reports or case series included in our study. We did not perform any extra assessment on the specialised image programming as done by Sakamoto et al.20 They devised a specialised image programming to exclusively study hypertelorism. This discripency in photographic adaptation of hypertelorism from literature has likely resulted in under-represention of a subset of hypertelorism in our case series. Hence direct comparison between both studies is inappropriate. Sakamoto et al20 reported cases of hypotelorism, of which nine had cyclopia. All were stillborn or died at an early age with severe holoprosencephaly. It is also worth mentioning the case of a neonate who survived with cyclopia with associated panophthalmitis.6
In our cohort, 35% (7 cases) of micro-ophthalmia/anophthalmia had bilateral involvement'. Ophthalmic abnormalities are not restricted to the same side of PL as the other side can also be affected. Biber21 in his review on PL quoted a case - featuring contralateral malformations. Guion-Almeida et al22 in a series of cerebro-ocular nares syndrome (CONS) defined a proboscis-like nare. Case number 9 in their series had ocular manifestations on another side of the proboscis-like nares. These observations highlight the possibility of contralateral involvement of ocular malformations as opposed to the popular belief of ipsilateral predilection in PL. The CONS series had higher frequency of brain abnormalities in cases with micro-ophthalmia/anophthalmia and hypertelorism. Including these cases in the present cohort may have influenced and skewed the association results.
In the present study, the associated anomalies in the brain (56.2%) and faces (54.7%) are higher compared with a study by English17 (19% and 38%, respectively). The Guion-Almeida et al study22 had documented serious brain abnormalities and profound facial oddity. These include a wide forehead, abnormal frontal hairline, high narrow palate and elongated philtrum.
Our study revealed a significant association between brain abnormalities or systemic abnormalities and hypertelorism and micro-ophthalmia/anophthalmia (p=0.000). To our interest, brain or systemic severity did not show an association with cumulative ocular abnormalities (p=0.249) or intraocular abnormalities (p=0.991). Hence, hypertelorism and micro-ophthalmia/anophthalmia reflect a diagnostic value. Their presence reinforces the need for neurological imaging and assessment. A positive association was noted between cumulative systemic abnormalities and cumulative ocular abnormalities (p=0.001). This suggests imploring a comprehensive ophthalmic evaluation in patients with systemic manifestations.
There are a few limitations to the present study. PL with other fatal malformations was excluded from this study. Such exclusion may have drawn a cohort of less affected subjects. The main potential for bias comes from the normal inference of unspecified ocular features. This may produce an understated prevalence of anomalies.
Conclusion
The results recommend an adequate ophthalmic evaluation while screening for a congenital deformity in cases of PL. Addressing PL in clinical practice needs a more comprehensive and multidisciplinary approach.
Ethics statements
Ethics approval
The study in question was not submitted to local ethical committee for approval since it involved a retrospective chart review of patients from existing literature and qualifies for exempt status under human subject regulations in our opinion. The study was conducted in accordance with Helsinki Declaration. However, for the representative case photograph shown in this article, due informed consent has been obtained and submitted.
References
Supplementary material
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors All authors included in the paper fulfil the criteria for authorship. AB conceptualised and supervised the study. ESS and PHS designed the computational framework and implementation. ESS wrote the manuscript with input from AS. BS performed the statistical data analysis. All authors discussed the results, provided critical feedback and structured the research, analysis and manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.